KEGG pathway maps¶
Objectives
Build a KEGG pathway map using R¶
In this exercise, we will generate KEGG a pathways map from genome annotations to visualize potential pathways present in our assembled, binned genome sequences.
The key package used here is pathview, available from Bioconductor (for installation on your local version of RStudio, see the previous intro to data presentation section). pathview is mainly used in transcriptomic analyses but can be adapted for any downstream analyses that utilise the KEGG database. For this exercise, we are interested in visualising the prevalence of genes that we have annotated in a pathway of interest.
To get started, if you're not already, log back in to NeSI's Jupyter hub and open RStudio.
1. Prepare environment¶
Set the working directory, load the required packages, and import data.
code
2. Subset the data¶
For this exercise, we will use KEGG Orthologies generated via DRAM. We'll start by loading the annotation file and extracting the relevant column.
code
3. Summarise KO per bin¶
Here, we are interested in the available KO in each bin. Thus, we can summarise the data by the bin to generate a list of KO per bin. Note that some annotations do not have KO numbers attached to them. In these cases, we will filter these data out.
Multiple KOs per bin
Multiple annotations per bin is possible and not entirely rare, even if you did filter by E-value/bitscore. Some genes may just be very difficult to tell apart based on pairwise sequence alignment annotations. In this case, we are looking for overall trends. Our question here is: Does this MAG have this pathway? We can further refine annotations by comparing domains and/or gene trees to known, characterised gene sequences if gene annotations look suspicious.
code
4. Identify pathway maps of interest¶
Before moving on, we must first identify the pathway map ID of our pathway of interest. Lets say, for this exercise, we are interested in the TCA cycle. Here, we will use KEGGREST to access the KEGG database and query it with a search term.
KEGGREST can also help you identify other information stored in the KEGG database. For more information, the KEGGREST vignette can be viewed using the vignette function in R: vignette("KEGGREST-vignette")
5. Reshaping the data for input into pathview¶
pathview needs the data as a numeric matrix with IDs as row names and samples/experiments/bins as column names. Here, we will reshape the data into a matrix of counts per KO number in each bin.
code
If you click on KO_matrix in the Environment pane, you can see that it is now a matrix of counts per KO per bin. Bins that do not possess a particular KO number is given NA. Do not worry about that as pathview can deal with that.
6. Creating pathway map of genes related to TCA cycle¶
Now we can generate images of the KEGG pathway maps using the matrix we just made. For this section, we will try to find genes invovled in the TCA cycle.
code
There is no plot output for this command as it automatically writes the results into the current working directory. By default, it names the file as <species><mapID>.<out.suffix>.png. If this is the first time this is run, it will also write the pathway map's original image file <species><mapID>.png and the <species><mapID>.xml with information about how the pathway is connected.
Lets take a look at our first output.
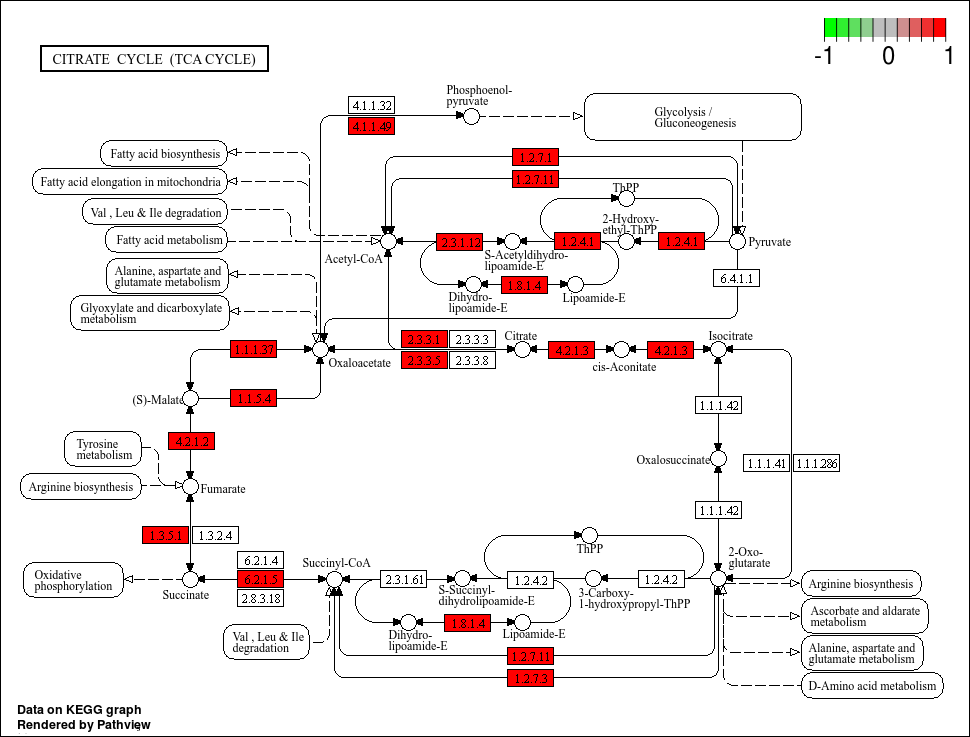
Boxes highlighted in red means that our MAG has this gene. However, the colour scale is a little strange seeing as there cannot be negative gene annotation hits (its either NA or larger than 0). Also, we know that there are definitely bins with more than 1 of some KO, but the colour highlights do not show that. Lets tweak the code further and perhaps pick better colours. For the latter, we will use the viridis colour package that is good for showing a gradient.
code
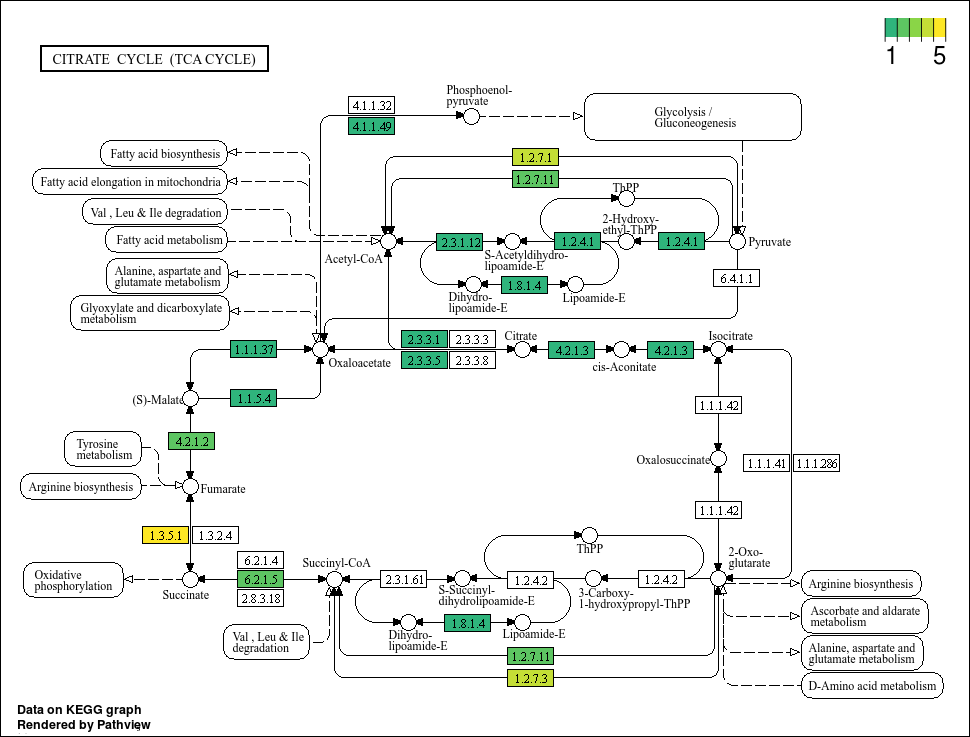
This plot looks much better. We can see that some genes do have more hits than others. Now, lets propagate this using map(...) based on our bin IDs.
code
Results

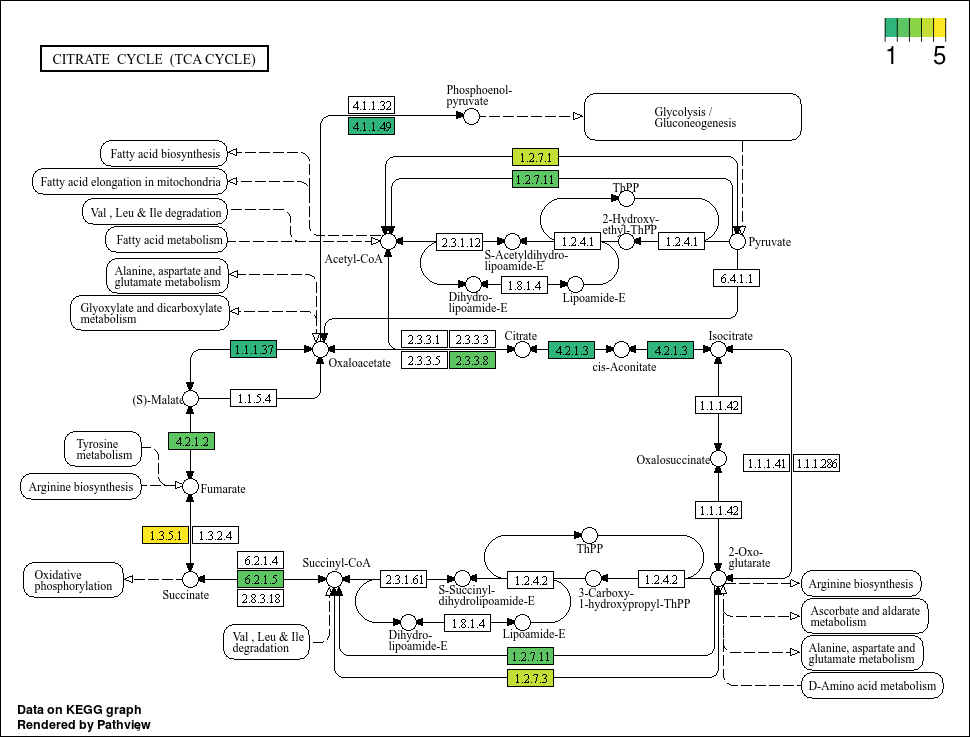
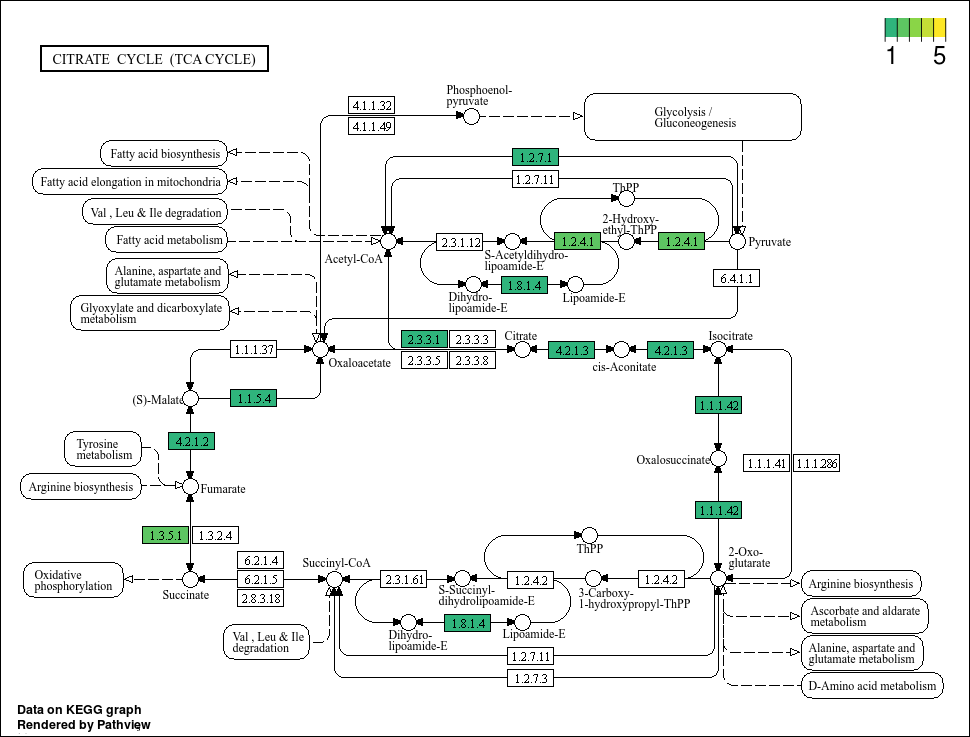
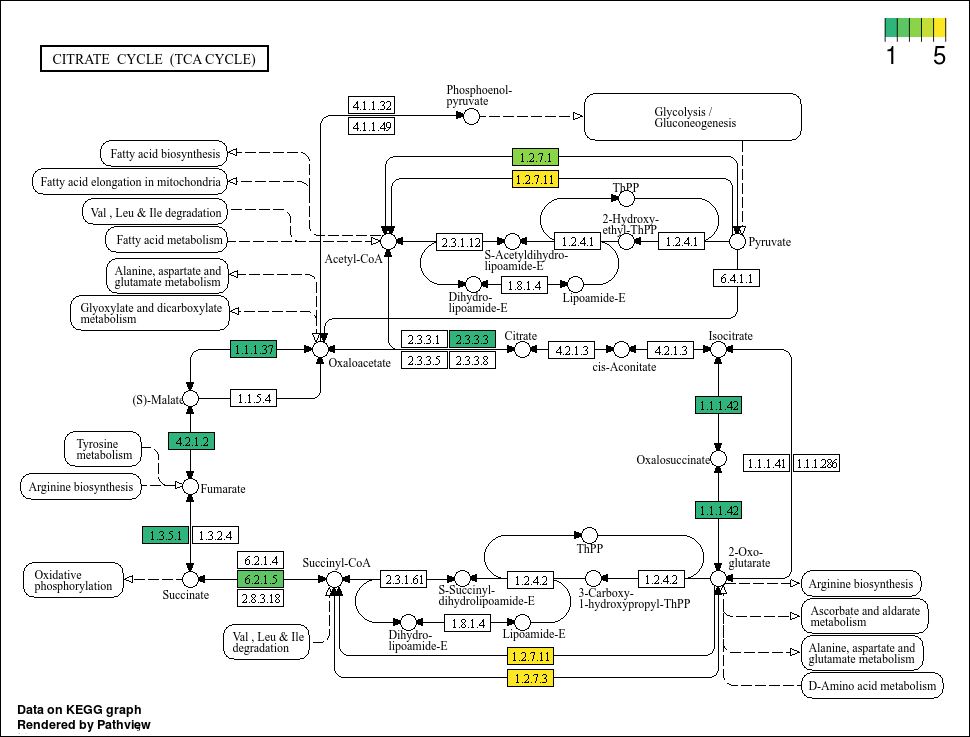


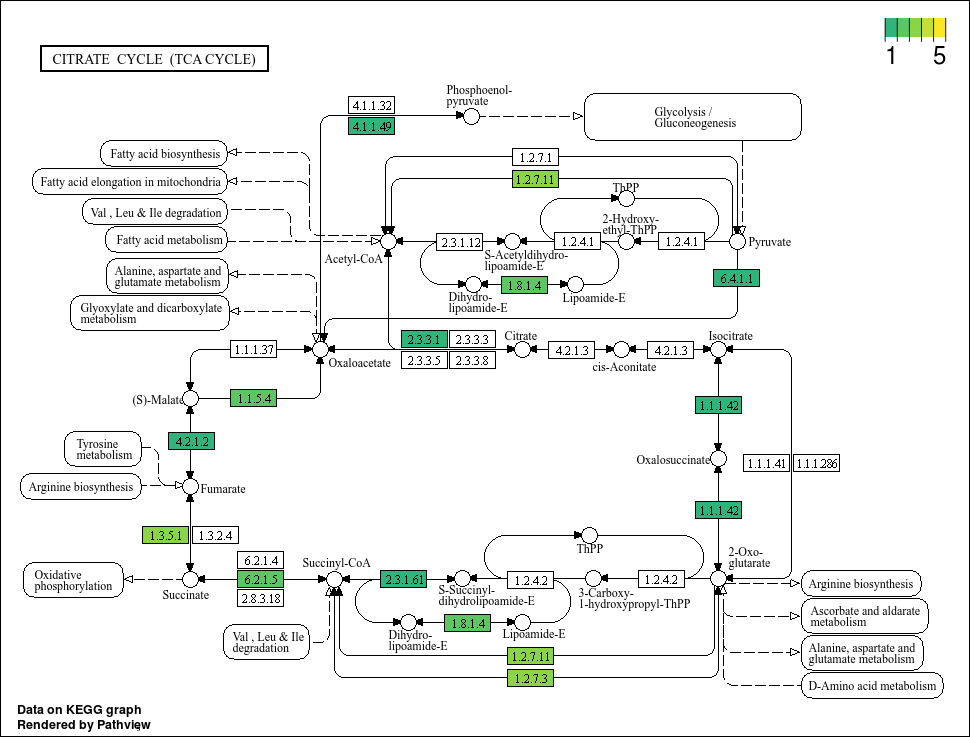
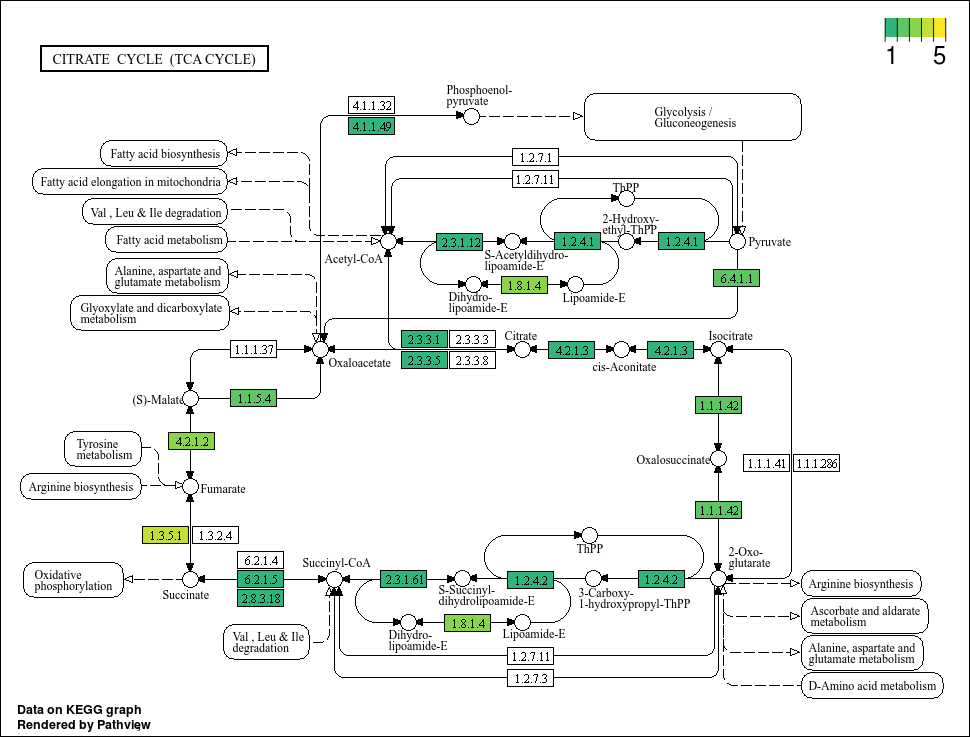
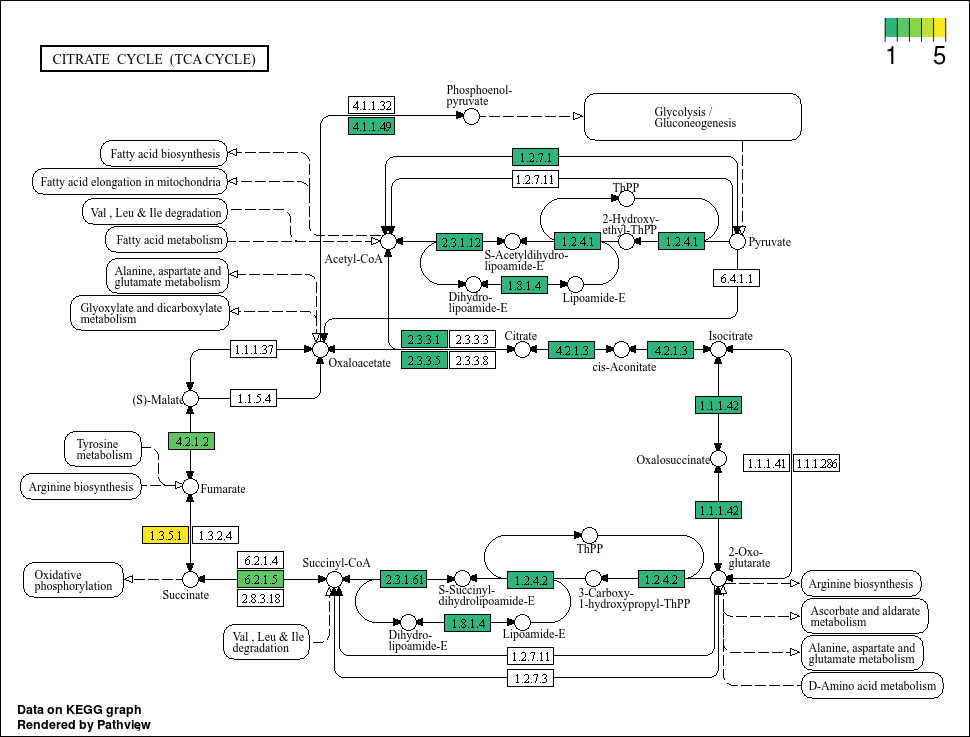
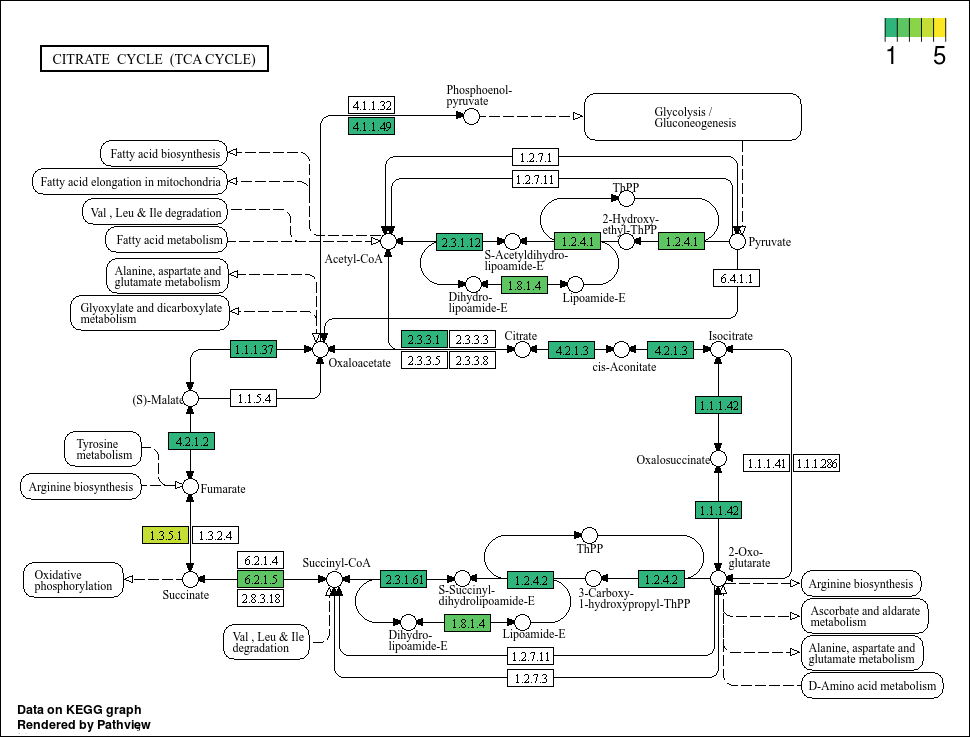
Based on the plots, it seems that not all bins have a complete TCA cycle.
Now that you know how to make pathway maps, try it using different pathways of interest!