APPENDIX (ex15): Generating pairwise contig comparisons using online BLAST¶
There are several ways of getting the BLAST files. genoplotR can read tabular files: either user-generated tab files (read_comparison_from_tab), or from BLAST output (read_comparison_from_blast). To produce files that are readable by genoPlotR, the -m 8 or 9 option should be used in blastall, or -outfmt 6 or 7 with the BLAST+ suite.
In this exercise, we are using tblastx on the NCBI website. Alternatively, you can use the command line version of tblastx in BLAST suite to get the same output (but remember to create the database first).
Firstly, we will need to get the input .fna files for blast. Navigate to the 11.data_presentation/gene_synteny/ folder, then we can grab the node of interest and load seqtk on Jupyter to grab the FASTA sequence.
code
cd /nesi/nobackup/nesi02659/MGSS_U/<YOUR FOLDER>/11.data_presentation/gene_synteny/
# Subset node name
for i in *cys.txt ;do
grep 'bin_' $i | sed 's/.*bin/bin/g;s/cov_\(.*\)_.*/cov_\1/g' | uniq > node_$i;
done
# Subset sequence using seqtk
export dir=/nesi/nobackup/nesi02659/MGSS_U/<YOUR FOLDER>/9.gene_prediction/filtered_bins/
module load seqtk
for i in {4,5,7};do
seqtk subseq ${dir}/bin_${i}.filtered.fna node_bin_${i}_cys.txt > bin_${i}_cys.fna;
done
Download the *cys.fna files to your local computer and then upload them to the NCBI website for blasting between bin 4 and bin 5, and then again between bin 5 and bin 7.
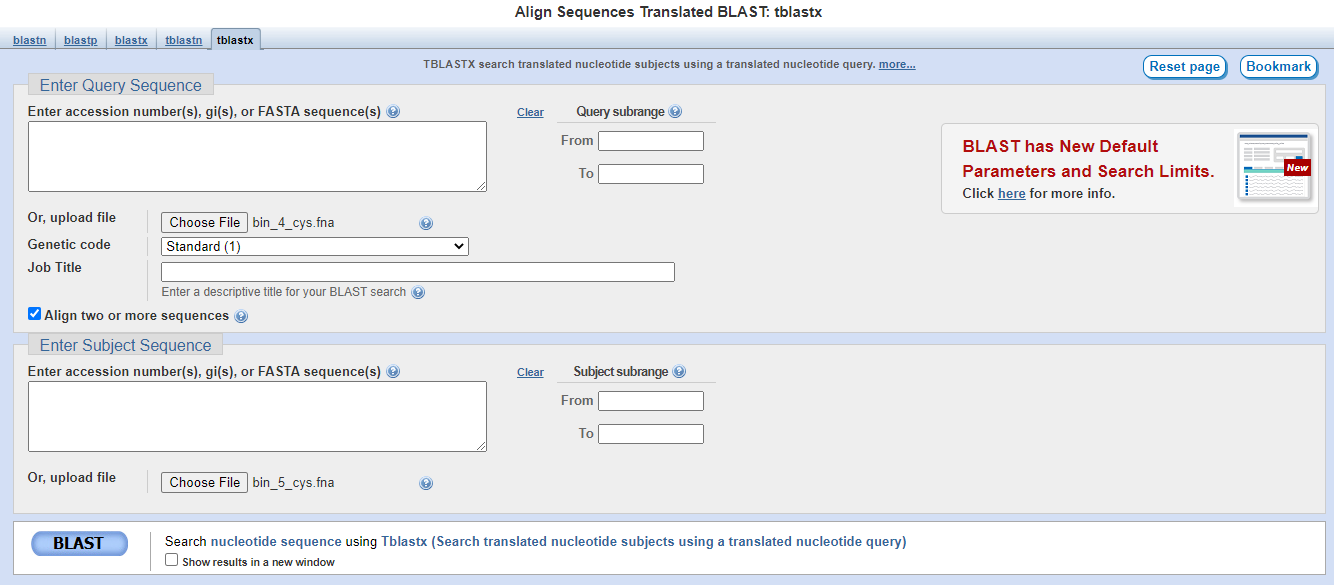
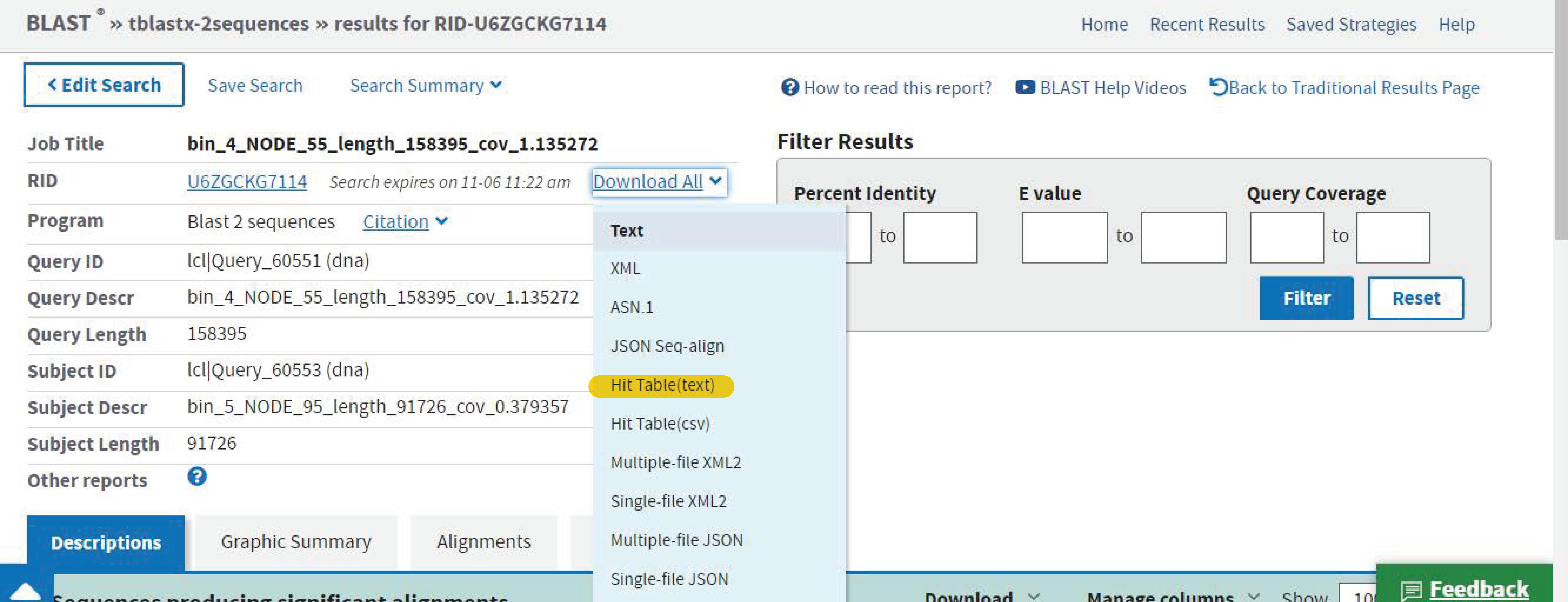
That's it! Now you will have downloaded two files (one comparing between bin 4 and bin 5, and another between bin 5 and bin 7).