Gene synteny¶
Build a sulfur assimilation gene alignment figure to investigate gene synteny using R¶
When investigating the evolution of genomes, we sometimes want to consider not only the presence/absence of genes in a genome, but also how they are arranged (e.g. in an operon). For this exercise, we are going to visualise several sulfur assimilation genes from our MAGs and compare their arrangements.
This exercise involves the use of commands in bash and R. Before starting, make sure you have an active terminal, then navigate to the directory 11.data_presentation/gene_synteny/. Here, you'll find a copy of the annotations that came from DRAM (dram_annotations.tsv) and a directory of dereplicated bins in dastool_bins/.
1. Obtain contigs containing genes of interest (R)¶
In an open RStudio session, set your working directory to 11.data_presentation/gene_synteny/ and then load the required libraries and annotation file.
code
In order to be able to assimilate sulfate, prokaryotic genomes need to encode for the following KEGG orthologies which encode for an ATP-dependent thiosulfate/sulfate uptake system:
- K02045 (cysA)
- K02046 (cysU)
- K02047 (cysW)
- K02048 (cysP) or K23163 (sbp)
We'll subset the annotation file to obtain relevant rows and columns. In addtion to the bin and KO information, we require the gene position relative to the contig, strandedness, and the start and end position of the gene.
code
2. Check contiguity of genes¶
We need to check that the genes are contiguous (all on the same contig) and sequential. If there gaps between genes, we will need to fill them.
code
Console output
We find that bin_7 is discontiguous. We need to identify how the genes of interest are distributed across the 2 contigs.
Console output
# A tibble: 5 × 3
# Groups: fasta [4]
fasta scaffold n_genes
<chr> <chr> <int>
1 bin_5 bin_5_NODE_55_length_158395_cov_1.135272 4
2 bin_7 bin_7_NODE_54_length_158668_cov_0.373555 1
3 bin_7 bin_7_NODE_96_length_91726_cov_0.379302 4
4 bin_8 bin_8_NODE_12_length_355954_cov_0.516970 3
5 bin_9 bin_9_NODE_62_length_149231_cov_0.651774 4
Let's also check which gene is not contiguous.
code
Console output
# A tibble: 5 × 3
scaffold gene_position ko_id
<chr> <dbl> <chr>
1 bin_7_NODE_54_length_158668_cov_0.373555 136 K02048
2 bin_7_NODE_96_length_91726_cov_0.379302 18 K02045
3 bin_7_NODE_96_length_91726_cov_0.379302 19 K02047
4 bin_7_NODE_96_length_91726_cov_0.379302 20 K02046
5 bin_7_NODE_96_length_91726_cov_0.379302 21 K23163
Looks like there is a an operon that consists of sbp and cysAUW, then a discontiguous cysP.
We also need to check for gaps between genes of interest.
Console output
# A tibble: 16 × 2
scaffold gene_position
<chr> <dbl>
1 bin_5_NODE_55_length_158395_cov_1.135272 128
2 bin_5_NODE_55_length_158395_cov_1.135272 132
3 bin_5_NODE_55_length_158395_cov_1.135272 133
4 bin_5_NODE_55_length_158395_cov_1.135272 134
5 bin_7_NODE_54_length_158668_cov_0.373555 136
6 bin_7_NODE_96_length_91726_cov_0.379302 18
7 bin_7_NODE_96_length_91726_cov_0.379302 19
8 bin_7_NODE_96_length_91726_cov_0.379302 20
9 bin_7_NODE_96_length_91726_cov_0.379302 21
10 bin_8_NODE_12_length_355954_cov_0.516970 148
11 bin_8_NODE_12_length_355954_cov_0.516970 149
12 bin_8_NODE_12_length_355954_cov_0.516970 150
13 bin_9_NODE_62_length_149231_cov_0.651774 55
14 bin_9_NODE_62_length_149231_cov_0.651774 56
15 bin_9_NODE_62_length_149231_cov_0.651774 57
16 bin_9_NODE_62_length_149231_cov_0.651774 58
Here, we can see there is a gap in bin_5 where we need to fill 3 genes.
3. Subset relevant genes and positions¶
Based on the explorations above, we need to:
- Add genes 129-131 for scaffold
bin_5_NODE_55_length_158395_cov_1.135272 - Remove
bin_7_NODE_54_length_158668_cov_0.373555
code
We will write out the results for processing in bash.
code
4. Subset relevant scaffolds from bin sequences¶
Now, we need to return to the terminal.
Check that you're in 11.data_presentation/gene_synteny/
We'll use the previously generated table information to extract the scaffolds (using seqtk) we need from bin files.
code
5. Compare scaffolds using tBLASTx¶
With our scaffolds ready, we can now perform pairwise comparisons between them. You can choose to generate complete combinations for comparisons. Here, we will only compare between bins sequentially. As we are using the -subject flag, the comparisons are single-threaded (we cannot change this).
code
-outfmt 7
You may be familiar with BLAST output format 6 as the tabular output from BLAST. Here, format 7 is the one with a commented header lines. This allows genoPlotR downstream to parse the columns appropriately. The proceeding arguments specifies a standard output std with additional columns for percentage of positive matches ppos and the translated frames for query/subject frames.
Now we have all the necessary files. We will return to the RStudio session for plotting.
6. Plot gene synteny¶
We first need to import all the relevant per bin files.
code
We will also create a data frame that has relevant information about the gene. In this case, we are using the gene symbol.
code
We then create tracks of DNA segments and annotations that can be parsed by genoPlotR.
code
Finally, we can plot!
code
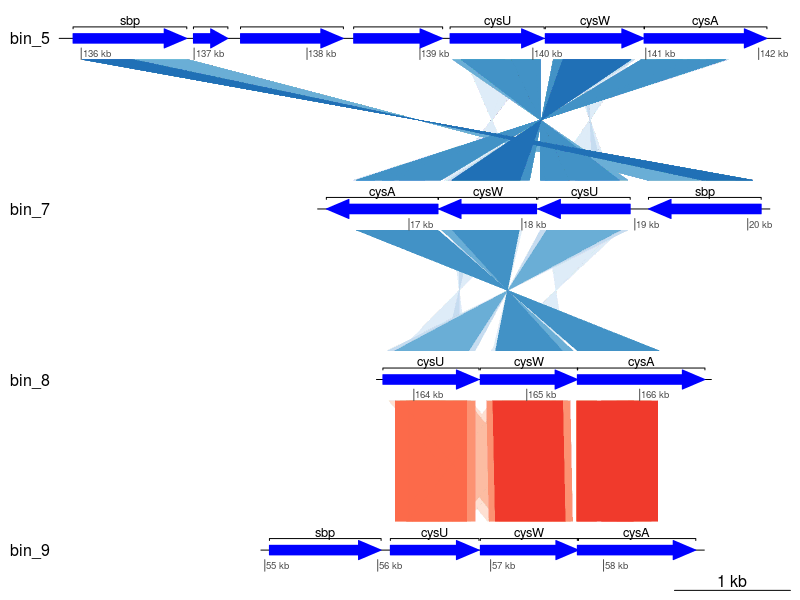
Seems a little messy, let's clean it up a little by only keeping those with the best matches (i.e., minimum E-value).
code
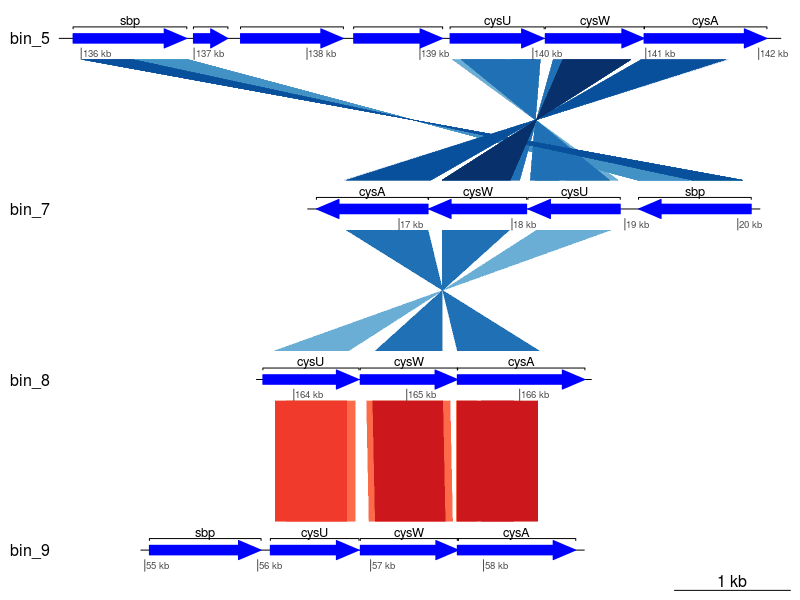
Much better! To further improve the plot, you might want to rearrange the tBLASTx comparisons and perhaps add additional information to the un-annotated genes based on hits to other databases (e.g., Pfam, TIGRFam, UniProt, etc.).