Introduction I: Shell¶
This lesson will be covered/referred during pre-Summer School sessions. We will start Day 1 with Introduction to HPC & HPC job scheduler
Objectives
Quick recap
- Log into the NeSI Jupyter service as per S.1.1 : NeSI Mahuika Jupyter login in NeSI Setup Supplementary material
- Then open a Jupyter Terminal Session
- We do recommend referring to NeSI File system,..Symlinks Supplementary material first
- This lesson is a quick recap on basic/essential linux commands and will be covered during the pre-summer school sessions.
- If you would like to follow through and learn a bit more on this topic, refer to Intermediate Shell for Bioinformatics material (or welcome to attend that workshop which will be offered two or three times per year)
Navigating your file system¶
-
Check the current working directory.(terminal session will be land on
/homedirectory) -
Switch to individual working directory on nobackup ( below )
-
OR you can navigate to above by using the symlink created as per instructions on Supplementary material with just
cd ~/mgss -
Change the directory to
MGSS_Intro -
Run the
lscommand to list the contents of the current directory. Check whether there are two .fastq files. -
The
mkdircommand (make directory) is used to make a directory. Entermkdirfollowed by a space, then the directory name you want to create
Copying, Moving, Renaming and Removing files¶
-
Make a second copy of
SRR097977.fastqand rename it asTest_1_backup.fastq.
-
Then move that file to
backup/directory. -
Navigate to
backup/directory and usemvcommand to rename and moveTest_1_backup.fastqasTest_1_copy.fastqto the directory immediately above. -
Return to the directory immediately above, check whether the
Test_1_copy.fastqwas moved and renamed as instructed and remove it by using thermcommand. -
See whether you can remove the
backup/directory by using thermcommand as well. -
By default,
rmwill not delete directories. This can be done by using-r(recursive) option.
Examining file contents¶
-
There are a number of ways to examine the content of a file.
catandlessare two commonly used programs for a quick look. Check the content ofSRR097977.fastqby using these commands. Take a note of the differences. -
A few useful shortcuts for navigating in
less
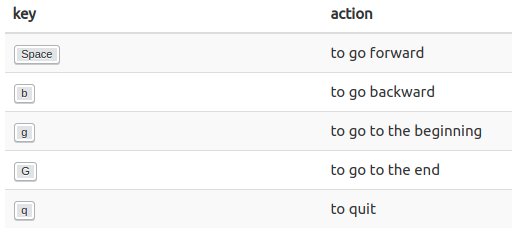
-
There are ways to take a look at parts of a file. For example, the
headandtailcommands will scan the beginning and end of a file, respectively. -
Adding
-noption to either of these commands will print the first or last n lines of a file.
Redirection and extraction¶
-
Although using
catandlesscommands will allow us to view the content of the whole file, most of the time we are in search of particular characters (strings) of interest, rather than the full content of the file. One of the most commonly used command-line utilities to search for strings isgrep. Let's use this command to search for the stringNNNNNNNNNNinSRR098026.fastqfile. -
Retrieve and discuss the output you get when
grepwas executed with the-B1and-A1flags. -
In both occasions, outputs were printed to the terminal where they can not be reproduced without the execution of the same command. In order for "string" of interest to be used for other operations, this has to be "redirected" (captured and written into a file). The command for redirecting output to a file is
>. Redirecting the string of bad reads that was searched using thegrepcommand to a file namedbad_reads.txtcan be done with -
Use the
wccommand to count the number of words, lines and characters in thebad_reads.txtfile. -
Add
-lflag towccommand and compare the number with the above output -
In an instance where the same operation has to be applied for multiple input files and the outputs are to be redirected to the same output file, it is important to make sure that the new output is not over-writing the previous output. This can be avoided with the use of
>>(append redirect) command which will append the new output to the end of the file, rather than overwriting it. -
Executing the same operation on multiple files with the same file extension (or different) can be done with wildcards, which are symbols or special characters that represent other characters. For an example. Using
*wildcard, we can run the previousgrepcommand on both files at the same time. -
The objective of the redirection example above is to search for a string in a set of files, write the output to a file, and then count the number of lines in that file. Generating output files for short routine tasks like this will end up generating an excessive number of files with little value. The
|(pipe) command is a commonly used method to apply an operation for an output without creating intermediate files. It takes the output generated by one command and uses it as the input to another command.
Text and file manipulation¶
There are a number of handy command line tools for working with text files and performing operations like selecting columns from a table or modifying text in a file stream. A few examples of these are below.
Cut¶
The cut command prints selected parts of lines from each file to standard output. It is basically a tool for selecting columns of text, delimited by a particular character. The tab character is the default delimiter that cut uses to determine what constitutes a field. If the columns in your file are delimited by another character, you can specify this using the -d parameter.
See what results you get from the file names.txt.
basename¶
basename is a function in UNIX that is helpful for removing a uniform part of a name from a list of files. In this case, we will use basename to remove the .fastq extension from the files that we've been working with.
sed¶
sed is a stream editor. A stream editor is used to perform basic text transformations on an input stream (a file, or input from a pipeline) like, searching, find and replace, insertion or deletion. The most common use of the sed command in UNIX is for substitution or for find and replace. By using sed you can edit files even without opening them, which is extremely important when working with large files.
- Some find and replace examples
Find and replace all chr to chromosome in the example.bed file and append the the edit to a new file named example_chromosome.bed
chr to chromosome, only if you also find 40 in the line
Find and replace directly on the input, but save an old version too
-i to edit files in-place instead of printing to standard output
- Print specific lines of the file
To print a specific line you can use the address function. Note that by default, sed will stream the entire file, so when you are interested in specific lines only, you will have to suppress this feature using the option -n
-n, --quiet, --silent = suppress automatic printing of pattern space
print 5th line of example.bed
We can provide any number of additional lines to print using -e option. Let's print line 2 and 5,
It also accepts range, using ,. Let's print line 2-6,
Loops¶
Loops are a common concept in most programming languages which allow us to execute commands repeatedly with ease. There are three basic loop constructs in bash scripting,
Types of Loops
iterates over a list of items and performs the given set of commands
For most of our uses, afor loop is sufficient for our needs, so that is what we will be focusing on for this exercise.
Shell identifies the for command and repeats a block of commands once for each item in a list. The for loop will take each item in the list (in order, one after the other), assign that item as the value of a variable, execute the commands between the do and done keywords, then proceed to the next item in the list and repeat over. The value of a variable is accessed by placing the $ character in front of the variable name. This will tell the interpreter to access the data stored within the variable, rather than the variable name. For example
This prevents the shell interpreter from treating i as a string or a command. The process is known as expanding the variable. We will now write a for loop to print the first two lines of our fastQ files:
for loops is basename which strips directory information and suffixes from file names (i.e. prints the filename name with any leading directory components removed).
basename is rather a powerful tool when used in a for loop. It enables the user to access just the file prefix which can be use to name things
Performs a given set of commands an unknown number of times as long as the given condition evaluates is true
Execute a given set of commands as longs as the given condition evaluates to false
Scripts¶
Executing operations that contain multiple lines/tasks or steps such as for loops via command line is rather inconvenient. For an example, imagine fixing a simple spelling mistake made somewhere in the middle of a for loop that was directly executed on the terminal.
The solution for this is the use of shell scripts, which are essentially a set of commands that you write into a text file and then run as a single command. In UNIX-like operating systems, inbuilt text editors such as nano, emacs, and vi provide the platforms to write scripts. For this workshop we will use nano to create a file named ForLoop.sh.
Add the following for-loop to the script (note the header #!/bin/bash).
Because nano is designed to work without a mouse for input, all commands you pass into the editor are done via keyboard shortcuts. You can save your changes by pressing Ctrl + O, then exit nano using Ctrl + X. If you try to exit without saving changes, you will get a prompt confirming whether or not you want to save before exiting, just like you would if you were working in Notepad or Word.
Now that you have saved your file, see if you can run the file by just typing the name of it (as you would for any command run off the terminal). You will notice the command written in the file will not be executed. The solution for this is to tell the machine what program to use to run the script.
Although the file contains enough information to be considered as a program itself, the operating system can not recognise it as a program. This is due to it's lacking "executable" permissions to be executed without the assistance of a third party. Run the ls -l ForLoop.sh command and evaluate the first part of the output
There are three file permission flags that a file we create on NeSI can possess. Two of these, the read (r) and write (w) are marked for the ForLoop.sh file .The third flag, executable (x) is not set. We want to change these permissions so that the file can be executed as a program. This can be done by using chmod command. Add the executable permissions (+x) to ForLoop.sh and run ls again to see what has changed.
Re-open the file in nano and append the output to TwoLines.txt, save and exit
Execute the file ForLoop.sh. We'll need to put ./ at the beginning so the computer knows to look here in this directory for the program.
Cheat sheet
ls- list the contents of the current directoryls -l- list the contents of the current directory in more detailpwd- show the location of the current directorycd DIR- change directory to directory DIR (DIR must be in your current directory - you should see its name when you typelsOR you need to specify either a full or relative path to DIR)cd -- change back to the last directory you were incd(alsocd ~/) - change to your home directorycd ..- change to the directory one level above
mv- move files or directoriescp- copy files or directoriesrm- delete files or directoriesmkdir- create a new directorycat- concatenate and print text files to screenmore- show contents of text files on screenless- cooler version ofmore. Allows searching (use/)tree- tree view of directory structurehead- view lines from the start of a filetail- view lines from the end of a filegrep- find patterns within files