{kind=link}
library(ggbeeswarm)
library(palmerpenguins)
library(tidyverse) Visualisation for ourselves - Exploratory Analysis
Visualisation is most often thought of as something we do for other people, to share some piece of knowledge we have. But equally important is the role visualisation can play in us learning something about our data. Visualisation can reveal trends or associations that we would otherwise struggle to identify in observations stored in rows and columns.
In this section we will perform some quick data visualisations that we can use for our own observations. The output of this section does not need to be overly attractive, but it does need to be clear and should ‘standalone’ (i.e., should speak for itself, without the need of accompanying notes).
The aims for this section are to give you the opportunity to practice some of the most important code used in visualisations and to force you to think about visualisation: what types of figures work well for certain data, how to display the information you want to convey.
We will examine one type of graph to represent four different types of data: distribution, correlation, parts of a whole, and evolution (change over time).
Distribution
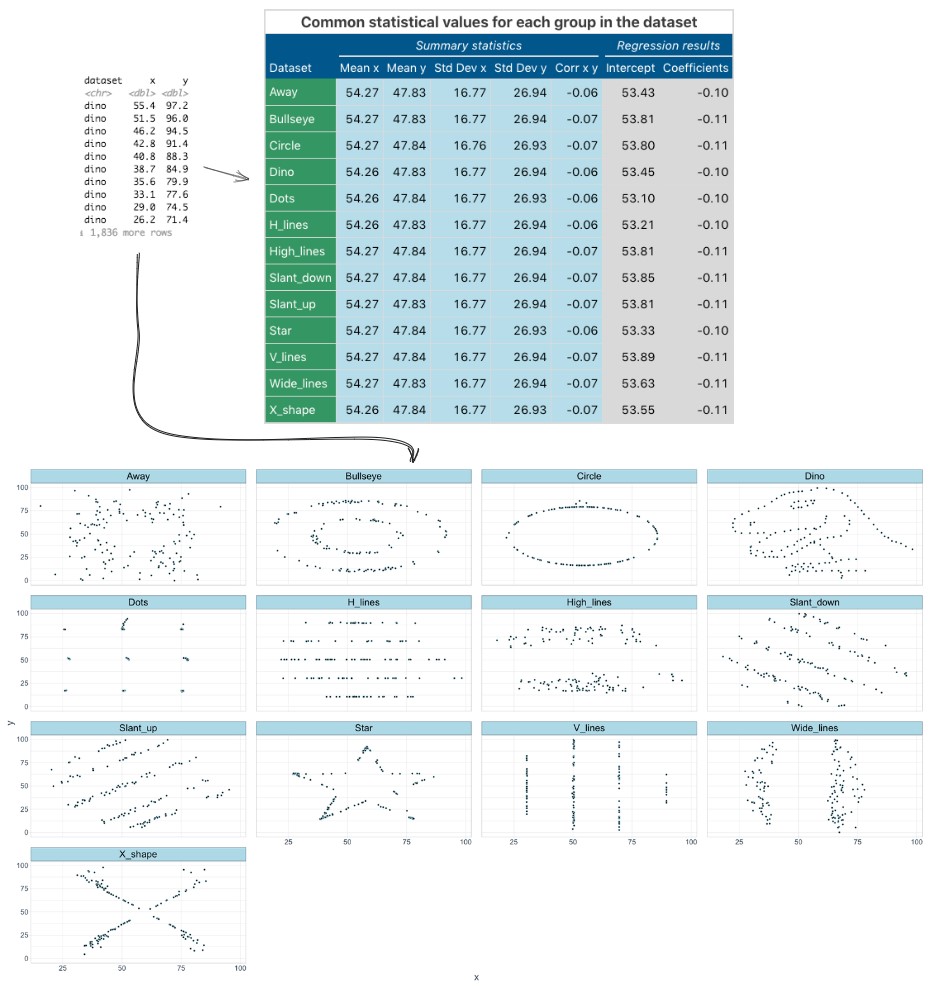
Distribution plots are a good place to start any data analysis, because many statistical tests rely on the assumption of a normal distribution. We may also make our own assumptions about how data is similar or different between different groups. The classic argument for distribution plots is the “datasaurus dozen” - a set of 13 datasets with identical summary statistics (same mean of x, mean of y, Std Dev etc.,) but when plotted reveal funny patterns (including points in the shape of a dinosaur).
Distribution matters!
Beeswarm plots with ggbeeswarm
Here we will use a type of plot, called a beeswarm plot, to highlight the power of visualisation.
Beeswarm plots can be made using a standalone package (install.packages("beeswarm")) or using a package that extends ggplot2 (install.packages("ggbeeswarm")). We will use the latter, since it lets us work within the ggplot format we have looked at already. This package has been pre-installed on the REANNZ training environment.
Load the ggbeeswarm and palmerpenguins packages. We will also use tidyverse later.
Beeswarm plots
Like with the geom_boxplot() we looked at in the previous episode, we need to specify where the data comes from and how the data is mapped. Beeswarm plots are simple and we will initially only need to specify the x and y axis data.
ggplot(data = penData,
mapping = aes(x = species,
y = flipper_length_mm)) +
geom_beeswarm()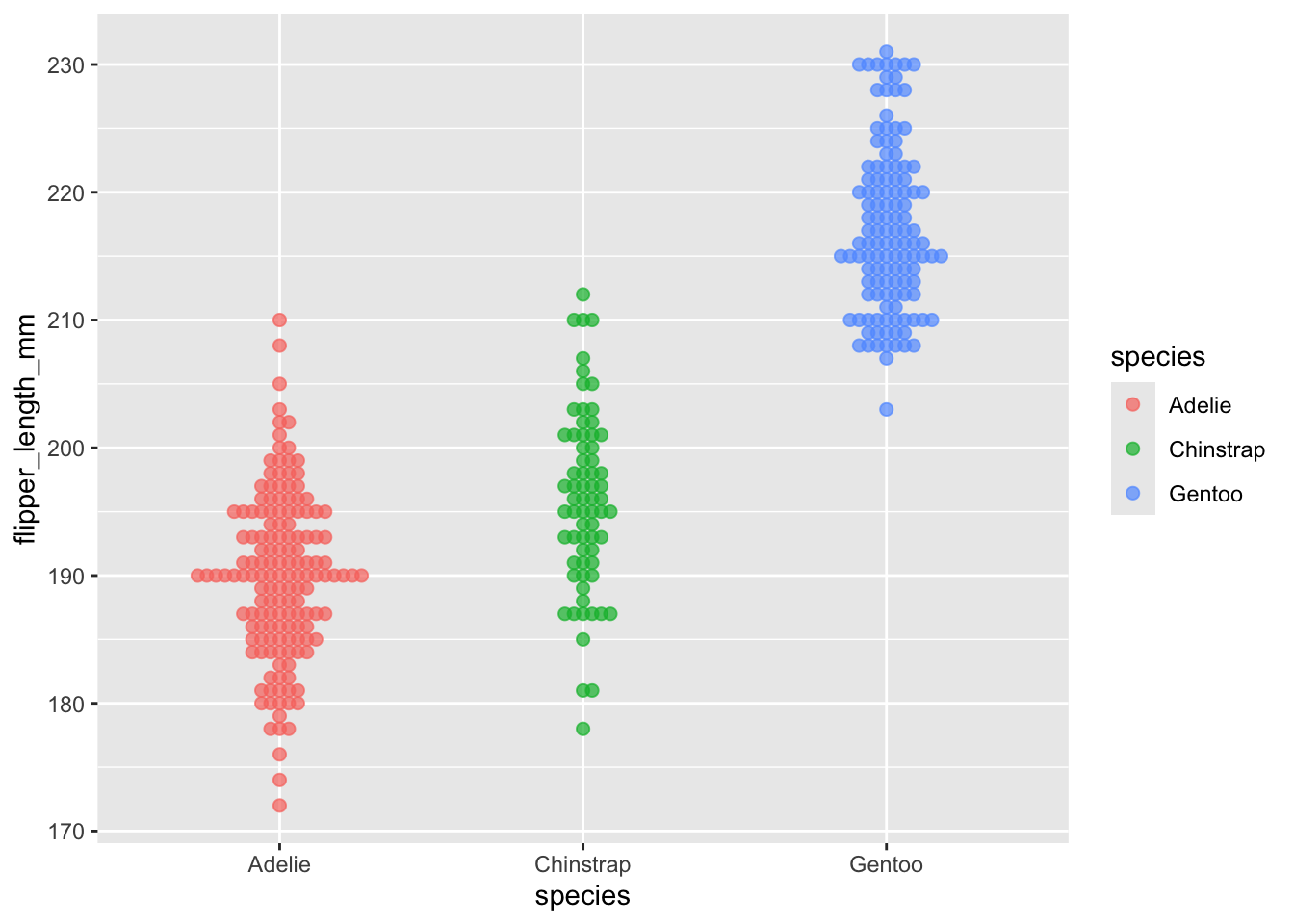
With this plain plot we can see what beeswarm is doing - when points on the y axis share a value they are moved horizontally so that overlapping points can be identified. This concept is commonly referred to as “jitter”, and “jitter plots” are a class of chart. Beeswarm plots are an implementation of jitter plots that aim to be compact with as little as possible overlap of points. This gives us the ability to see density through the width of the plot, similar to how a violin plot (geom_violin) works.
Let’s add in the violin plot as another geom layer to further visualise our distribution. At the same time, we’ll add in some more arguments for adjusting the visualisation.
Distribution plots are a good opportunity to introduce two new parameters we can adjust in ggplot: size and alpha.
Size is simply the size of the data point. We can assign a single value for all points, or can assign a continuous variable.
Alpha is the level of transparency. When data points are too large (from the size argument), or too numerous, the points can overlap one another (a problem called “overplotting”). This is particularly problematic outside of beeswarm. When we set alpha to something less than 1, the points become somewhat transparent. Therefore, if two points overlap one another, it is clear from the change in colour (as two transparent but overlapping points are darker than a single point).
ggplot(data = penData,
mapping = aes(x = species,
y = flipper_length_mm)) +
geom_beeswarm(size = 2) + # changes the point size
geom_violin(alpha = 0.4) # makes the violin layer 40% opaque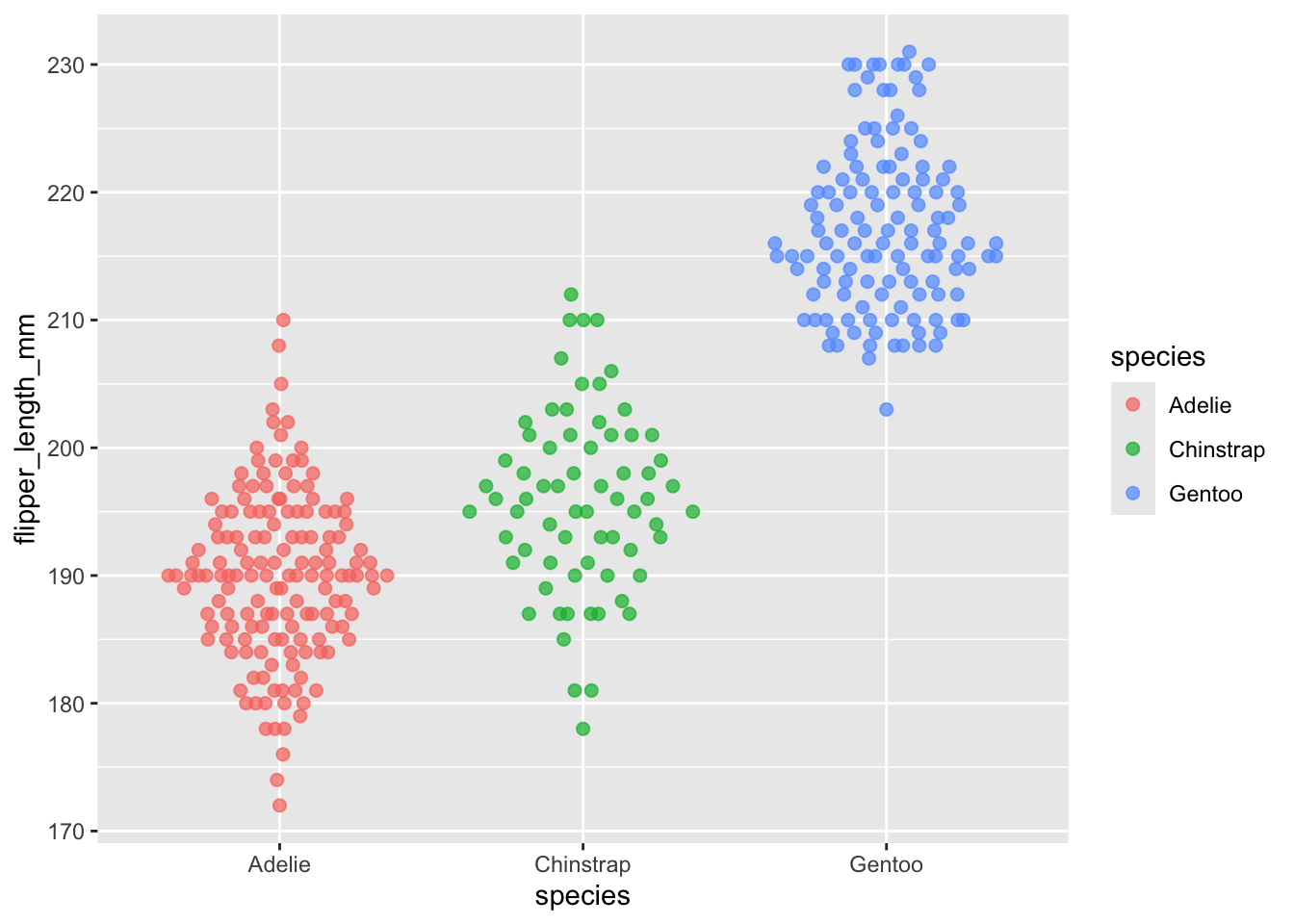
EXERCISE 🧠🏋️♀️ (1 min)
ggplot2 builds up geoms in layers, therefore the geom_beeswarm layer is plotted first, then the geom_violin layer is plotted on top.
Try the following and see how the plot changes:
- Change the
alpha = 0.4argument in geom_violin toalpha = 1andalpha = 0 - Swap the order of the two geoms, so that
geom_violinis plotted first andgeom_beeswarmsecond.
DISCUSSION 🤔 (3 mins)
Did you notice anything unusual about the distribution story being told by our geom_beeswarm and geom_violin layers?
The violin for Chinstrap in particular appears wide in the middle, yet the beeswarm points suggest there aren’t many individuals with flipper lengths in that range. This mismatch happens because geom_violin draws a smoothed curve that can create the impression of more density than what the raw points support, especially with smaller samples.
By default, geom_violin uses scale = "area", which gives every violin the same total area regardless of how many observations it represents. This is reasonable when group sizes are similar and you want to compare the shape of distributions. But when group sizes differ:
table(penData$species)
Adelie Chinstrap Gentoo
146 68 119 …it can be misleading, because a violin based on 68 observations looks just as “confident” as one based on 146. We can address this with scale = "count", which scales violin width proportionally to sample size:
ggplot(data = penData,
mapping = aes(x = species,
y = flipper_length_mm)) +
geom_beeswarm(size = 2) +
geom_violin(alpha = 0.4, scale = "count") # default is scale = "area" 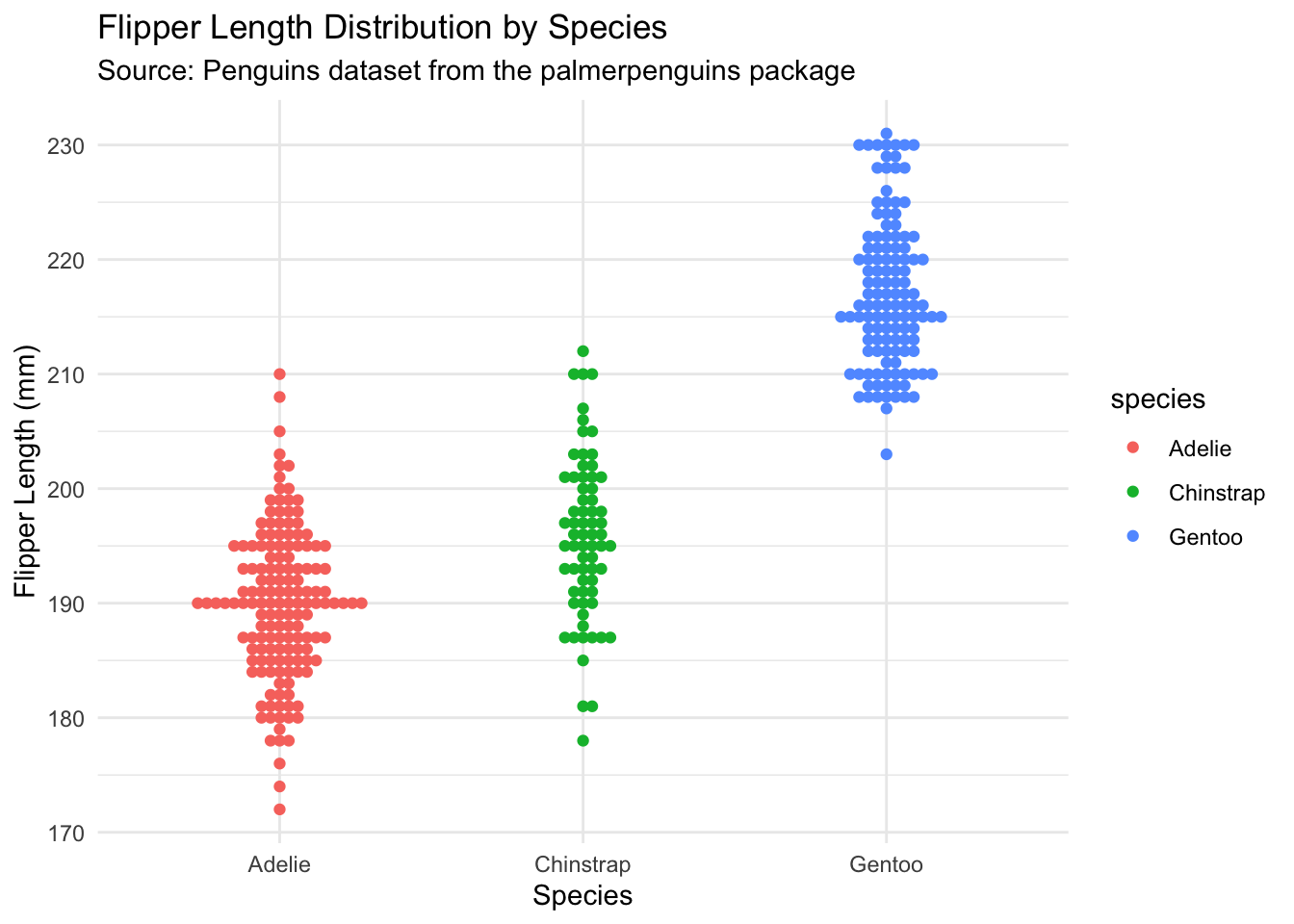
With scale = "count", the Chinstrap violin is visibly narrower than the others, visually demonstrating that this group has fewer observations and its distribution shape should be interpreted with more caution.
This gives us a more realistic view of our distribution in the context of the count i.e., the number of samples per group. It is not necessarily wrong to use one or the other scale for violin plot – you need to decide as the researcher how to best represent the story your data to tells!
Jittering the points with geom_jitter()
A commonly used function for visualising data points that overlap is geom_jitter(). “Jittering” adds a small amount of random variation to each point to reduce overplotting. The beeswarm plot is similar in that it reduces overplotting (i.e., overlapping points), but it does so in a more structured way which creates a compact visualisation. True jittering, like the geom_jitter() adds random variation.
EXERCISE 🧠🏋️♀️ (1 min)
Using the code below as a base, add geom_jitter() and compare the output to our earlier plots using beeswarm and violin. Which geom do you prefer and which one better represents the distribution of the data?
Optional: change size and alpha arguments in your geom_jitter() too.
ggplot(data = penData,
mapping = aes(x = species,
y = flipper_length_mm,
colour = species))
SOLUTION
ggplot(data = penData,
mapping = aes(x = species,
y = flipper_length_mm,
colour = species)) +
geom_jitter(size = 2,
alpha = 0.7)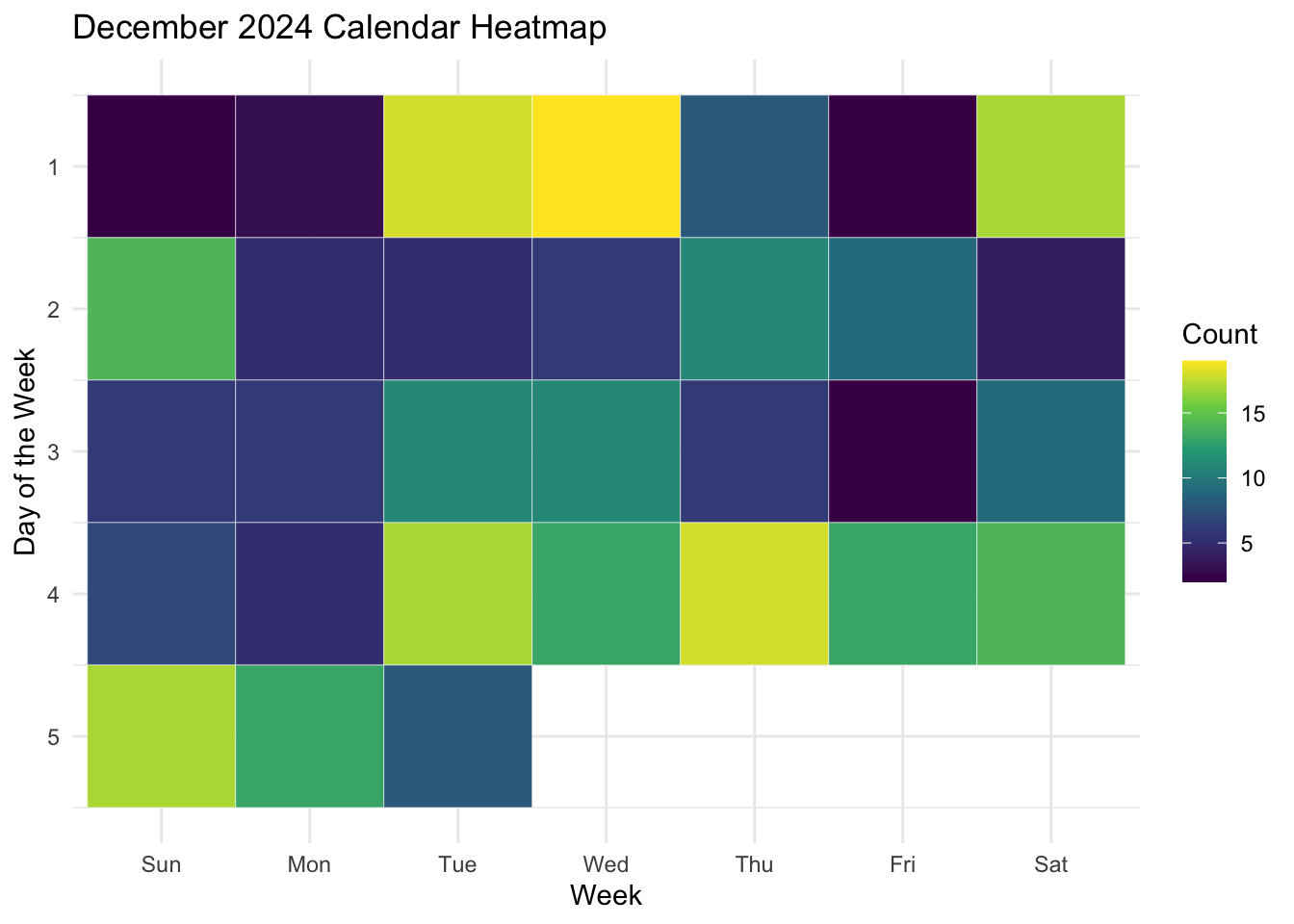
Looks like we have lost a lot of the distribution information here, that we could see more clearly in the beeswarm plot. This is a good example of how the wrong choice of plot can lead to a misleading visualisation.
While useful for showing individual observations, jitter does not meaningfully represent distribution shape and can sometimes obscure density patterns. For visualising distributions, approaches such as beeswarm, violin, or boxplots are often more appropriate.
Note: because the variation is random, everytime you generate this plot it will look slightly different! You can make the plot reproducible using set.seed() – we’ll do this in a later examples too.
Theme and labels
We can add two other functions to make this plot much easier to read. First, the theme_minimal() function is a quick way to set a global theme to the plot and make it less cluttered. The second function is labs(), which we use to add a title, subtitle, caption and x and y axes labels.
ggplot(data = penData,
mapping = aes(x = species,
y = flipper_length_mm,
colour = species)) +
geom_beeswarm() +
theme_minimal() +
labs(title = "Flipper Length Distribution by Species",
subtitle = "Source: Penguins dataset from the palmerpenguins package",
x = "Species",
y = "Flipper Length (mm)")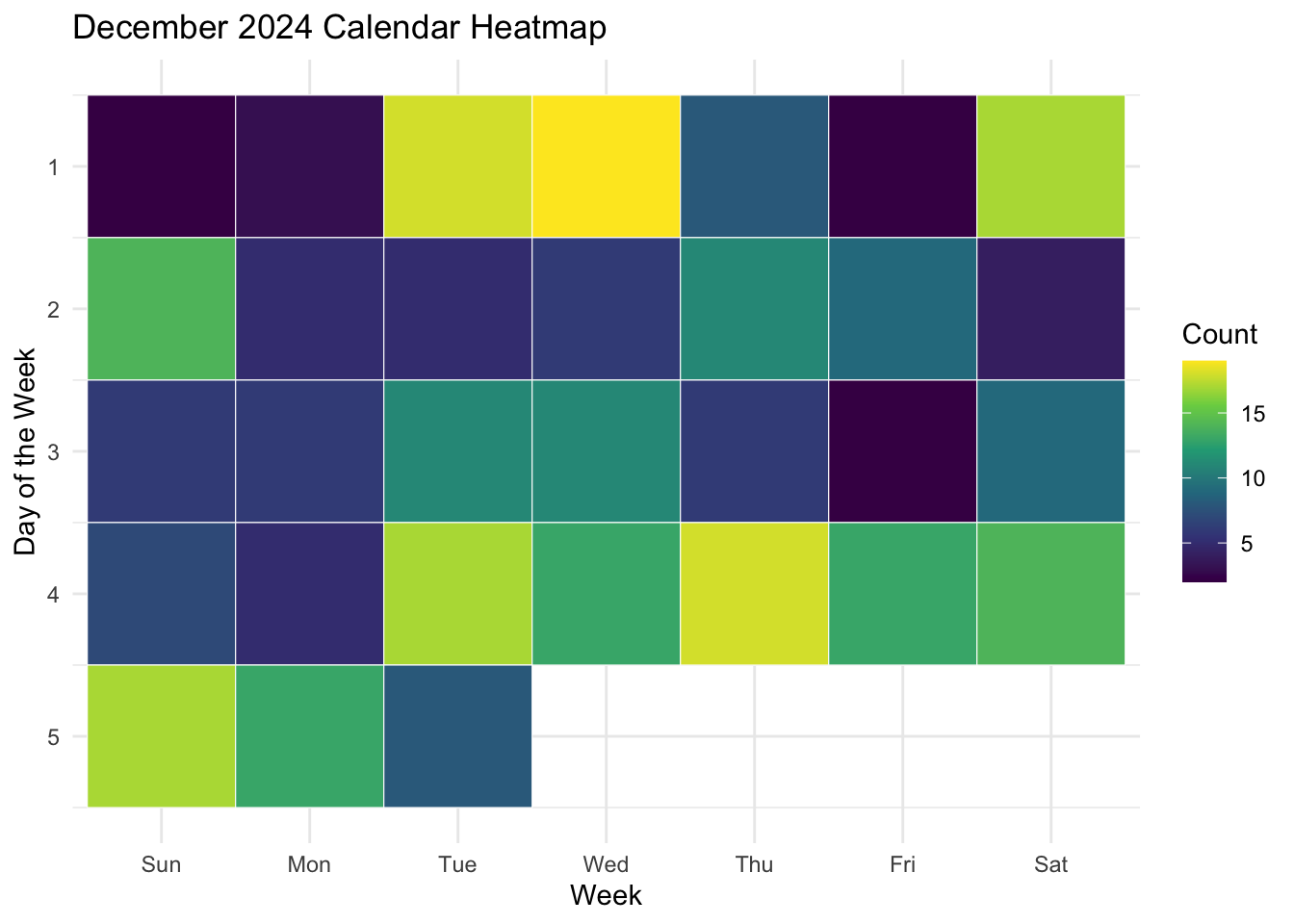
EXERCISE 🧠🏋️♀️ (1 min)
Have a play with different themes, which one do you like best for this plot? Can you find even more suggested themes in RStudio that you like better?
theme_minimal()theme_classic()theme_bw()
We will add theme_minimal() to every plot we do today to keep them looking tidy and coherent.
Correlation
Heatmaps with geom_tile()
Heatmaps can be used to visualise values across two dimensions using colour. In a biological context this could be gene expression values across a range of genes for a set of samples. This is often combined with some type of clustering (e.g., unsupervised hierarchical clustering with k-means) to identify patterns of gene expression which differ between sample groups.
Here we will use geom_tile() to create a particular type of heatmap called a “calendar heatmap”, which will display a value on a given day for a selected time period. The example below is captured from github, which display the number of commits (saves) a user made across the year.
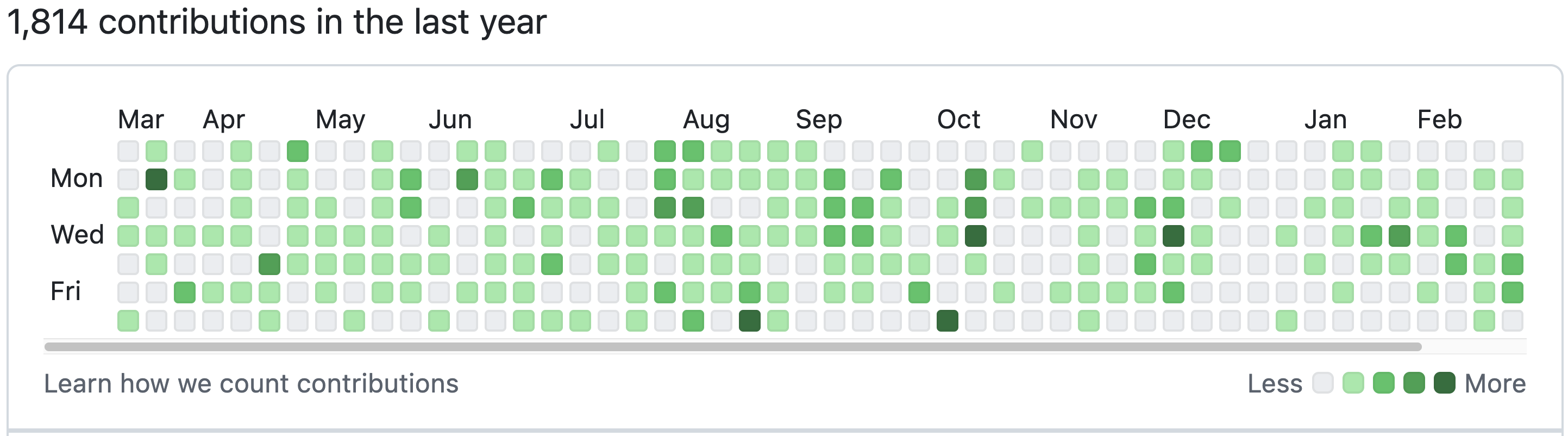
That’s a lot of commits!
Choose your own adventure 👣:
1. Calendar heatmap example
Calendar heatmap
We will create a simple calendar heatmap to demonstrate the concept. Remember, the goal here is to expose you to different types of plots and for you to practice using ggplot2.
Generating example data
First, we will generate some example data. Copy and paste this code, because it is not central to our understanding:
set.seed(0982)
month_data <- tibble(
date = seq(as.Date("2024-12-01"),
as.Date("2024-12-31"),
by = "day"),
count = sample(1:20, 31, replace = TRUE) # Random counts per day
) |>
mutate(
weekday = wday(date, label = TRUE, abbr = TRUE), # Day of the week (Sun-Sat)
week = (day(date) - 1) %/% 7 + 1 # Week number (1 to 5)
)
month_data |> head() # A tibble: 6 × 4
date count weekday week
<date> <int> <ord> <dbl>
1 2024-12-01 2 Sun 1
2 2024-12-02 3 Mon 1
3 2024-12-03 18 Tue 1
4 2024-12-04 19 Wed 1
5 2024-12-05 8 Thu 1
6 2024-12-06 2 Fri 1geom_tile()
Now we will create the plot with geom_tile(). Like with the geom_boxplot() we looked at in the previous episode, we need to specify where the data comes from and how the data is mapped. With geom_tile() we will always need to use aes() to specify the x and y, and we will also specify the variable that is used to determine the colour of each tile, which we do with “fill =”. In our example we will map the count variable to fill.
ggplot(data = month_data,
mapping = aes(x = week,
y = weekday,
fill = count)) +
geom_tile()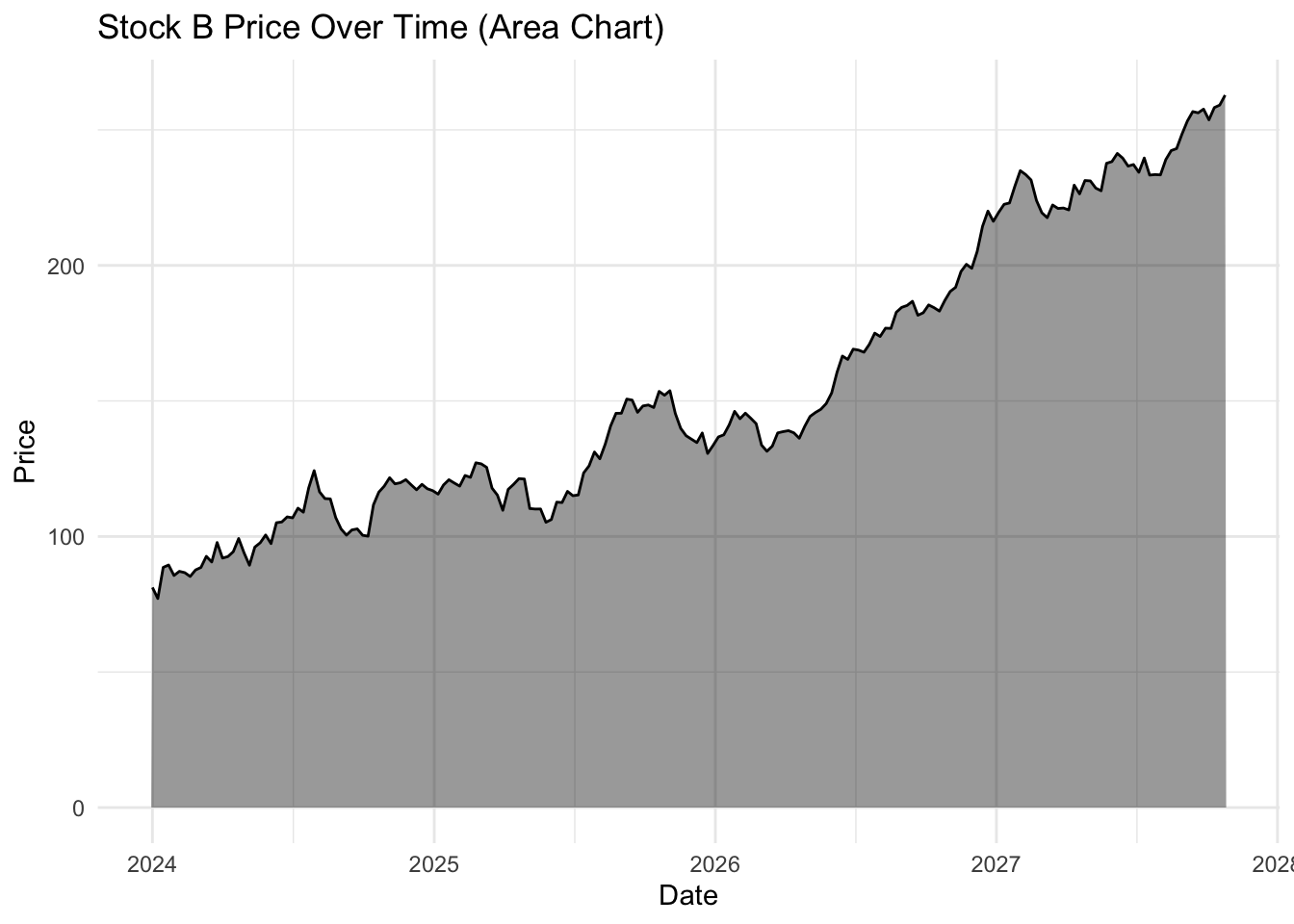
This is a good starting point in that we have functionally sketched the data into the plot type. However, we can see that it the layout does not match our expectations for a calendar: it reads the days in reverse (Saturday, Friday, Thursday etc.,), and has the week on the x axis. It is also missing a title, and axes labels, and it doesn’t have particularly high contrast between high and low values.
To correct these issues we will:
Swap the x and y axis with the
aes()function.Use the
scale_y_reverse()function to reverse the Y-axis (which will now be weeks) so that the last value (week 5) is placed at the bottom.Add the
scale_fill_viridis_c()function to change the colour of the fill. The “scale_[argument]_viridis” set of functions are used to select a colour blind-friendly pallette with high contrast.Use the
labs()function to add title, x and y axis labels, and rename the fill (legend) variable.Additionally, we will add colour = “white” as an argument for
geom_tile(), which will result in a fine white grid separating our tiles, and add thetheme_minimal()function to reduce the visual clutter of the grey background in the plot.
Exercise: Use the code block and tips above to create an improved version of the calendar heatmap.
You do not need to do all of these changes, but try them out if you can! Remember to add a “+” at the end of each line when adding new functions.
Solution
ggplot(data = month_data,
mapping = aes(x = weekday,
y = week,
fill = count)) +
geom_tile(colour = "white") +
scale_fill_viridis_c() +
scale_y_reverse() +
theme_minimal() +
labs(title = "December 2024 Calendar Heatmap",
x = "Week",
y = "Day of the Week",
fill = "Count")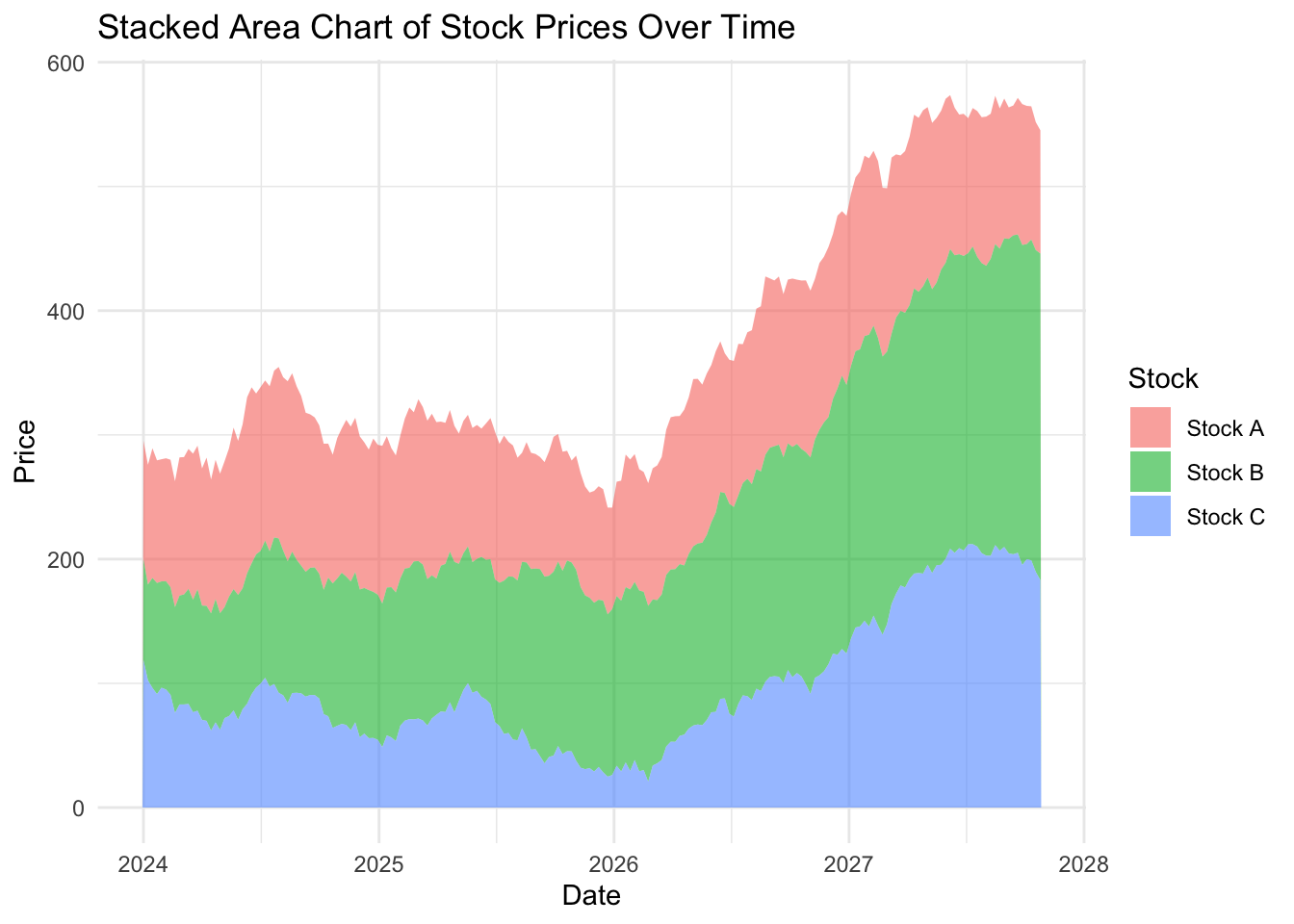
2. Gene expression heatmap example
Gene expression heatmap
We will create a simple gene expression heatmap to demonstrate the concept.
Generating example data
First, we will generate some example data. Copy and paste this code, because it is not central to our understanding:
set.seed(982)
# 20 genes across 6 samples
expression_data <- expand_grid(
gene = paste0("Gene_", 1:20),
sample = paste0("Sample_", 1:6)
) |>
mutate(
expression = rnorm(n(), mean = 10, sd = 2)
)
expression_data |> head()# A tibble: 6 × 3
gene sample expression
<chr> <chr> <dbl>
1 Gene_1 Sample_1 8.98
2 Gene_1 Sample_2 7.75
3 Gene_1 Sample_3 13.0
4 Gene_1 Sample_4 9.90
5 Gene_1 Sample_5 8.42
6 Gene_1 Sample_6 10.6 Note: This format the data is in is called “long format”, which is the format that ggplot2 is designed to work with. If you have data in “wide format” (where each sample is a column), you can use the pivot_longer() function from the tidyverse to convert it to long format. See this Long vs wide data blog post here or check out our Supplementary page on data manipulation.
geom_tile()
Now we will create the plot with geom_tile(). Like with the geom_boxplot() we looked at in the previous episode, we need to specify where the data comes from and how the data is mapped. With geom_tile() we will always need to use aes() to specify the x and y, and we will also specify the variable that is used to determine the colour of each tile, which we do with “fill =”. In our example we will map the count variable to fill.
ggplot(expression_data,
aes(x = sample,
y = gene,
fill = expression)) +
geom_tile()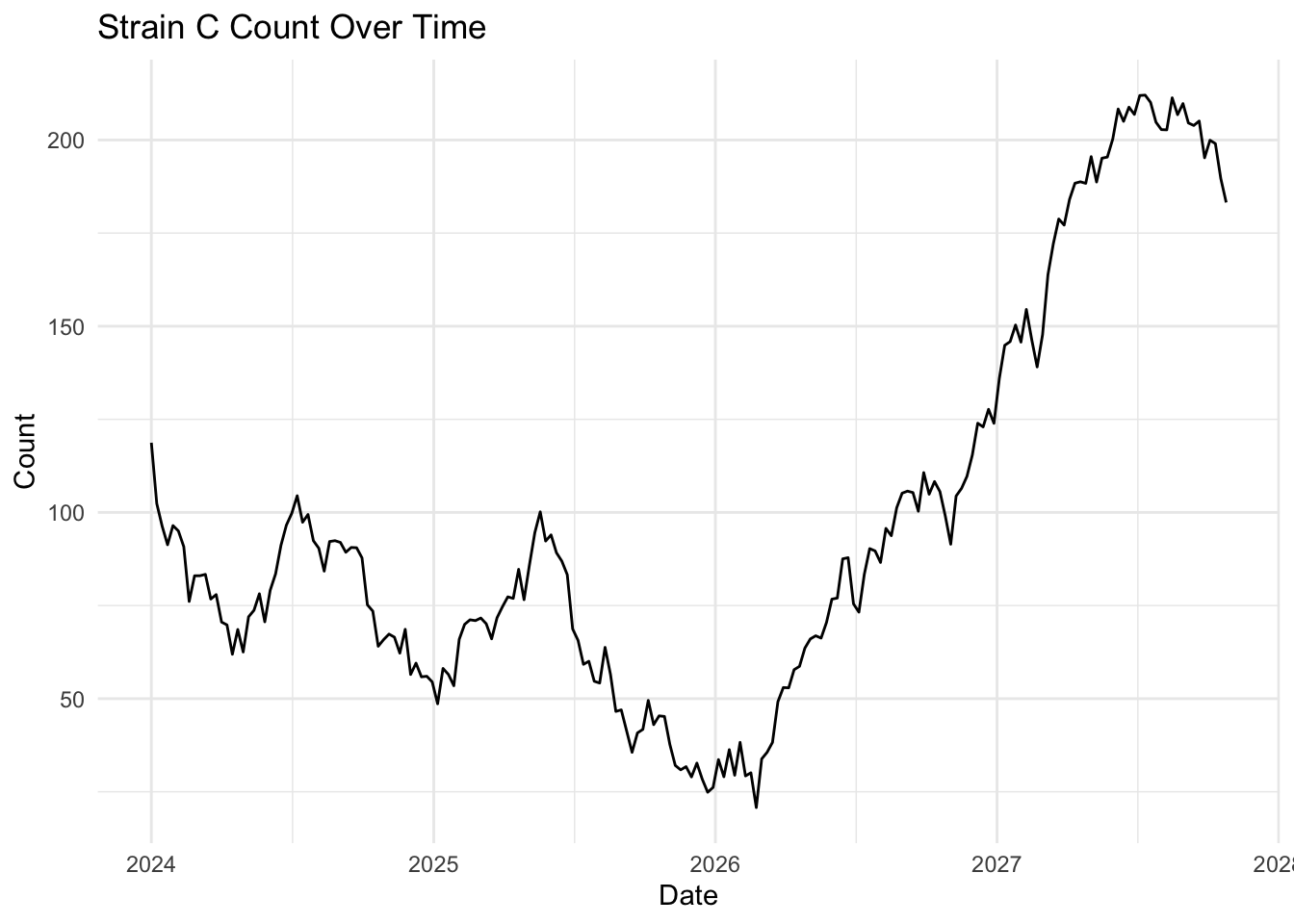
This is a good starting point in that we have functionally sketched the data into the plot type. However, we can see that it the layout does not match our expectations for a gene expression heatmap: it has no title, and axes labels, and it doesn’t have particularly high contrast between high and low values.
EXERCISE 🧠🏋️♀️ (5 mins)
To correct these issues we will:
- Add the
scale_fill_viridis_c()function to change the colour of the fill.
- Use the
labs()function to add title, x and y axis labels, and rename the fill (legend) variable.
- Additionally, we will add colour = “white” as an argument for
geom_tile(), which will result in a fine white grid separating our tiles, and add thetheme_minimal()function to reduce the visual clutter of the grey background in the plot.
Solution
ggplot(expression_data,
aes(x = sample,
y = gene,
fill = expression)) +
geom_tile(colour = "white") +
scale_fill_viridis_c() + #Alternatively, you could use scale_fill_gradient(low = "blue", high = "red") for a different colour scheme
theme_minimal() +
labs(title = "Gene Expression Heatmap",
x = "Sample",
y = "Gene",
fill = "Expression (log2 counts)") 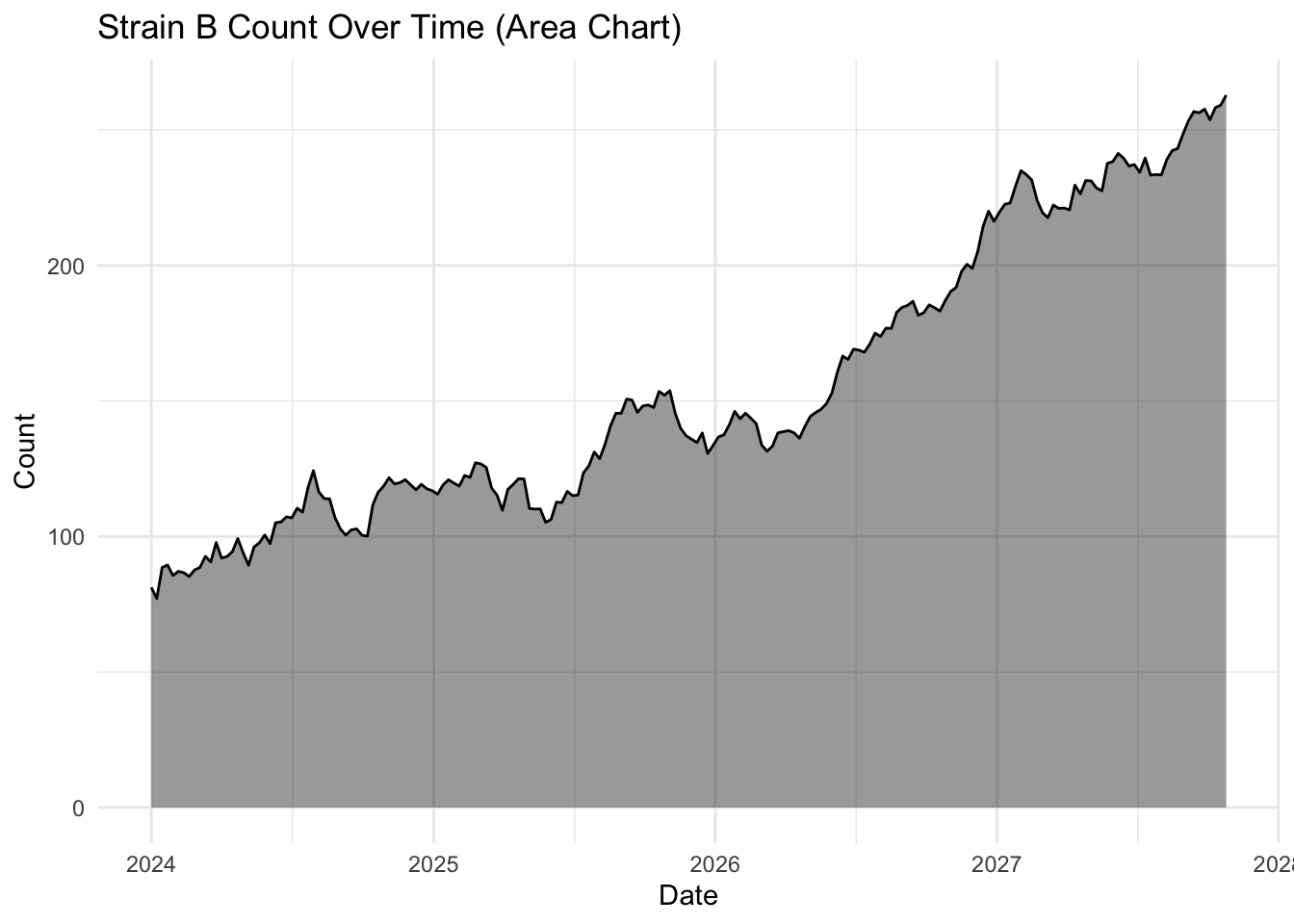
Note: gene expression data needs to be transformed (e.g., log2 transformed) before plotting. This is covered in more detail in our RNA-seq Data Analysis workshop.
Another way to plot heatmaps for gene expression data is with the pheatmap package, which has a lot of built in functionality for clustering and annotation. Specifically it allows you to to easily include a dendrogram to show the results of hierarchical clustering, which is a common way to visualise patterns in gene expression data.
See here R documentation on pheatmap package.
Parts of a whole
A simple example of a ‘Parts of a whole’ graph is the stacked bar plot. Let’s do an example of one here using our palmer penguins dataset again, then take a break from executing code and look at some examples.
To create a stacked bar plot, we set the x-axis as our categorical variable and the fill as the variable we want to see proportions for. We don’t need to set the y-axis variable as it will default to the count of our fill. We also don’t need to use a new geom for this plot – we use geom_bar().
ggplot(penData, aes(x = species, fill = sex)) +
geom_bar() +
theme_classic()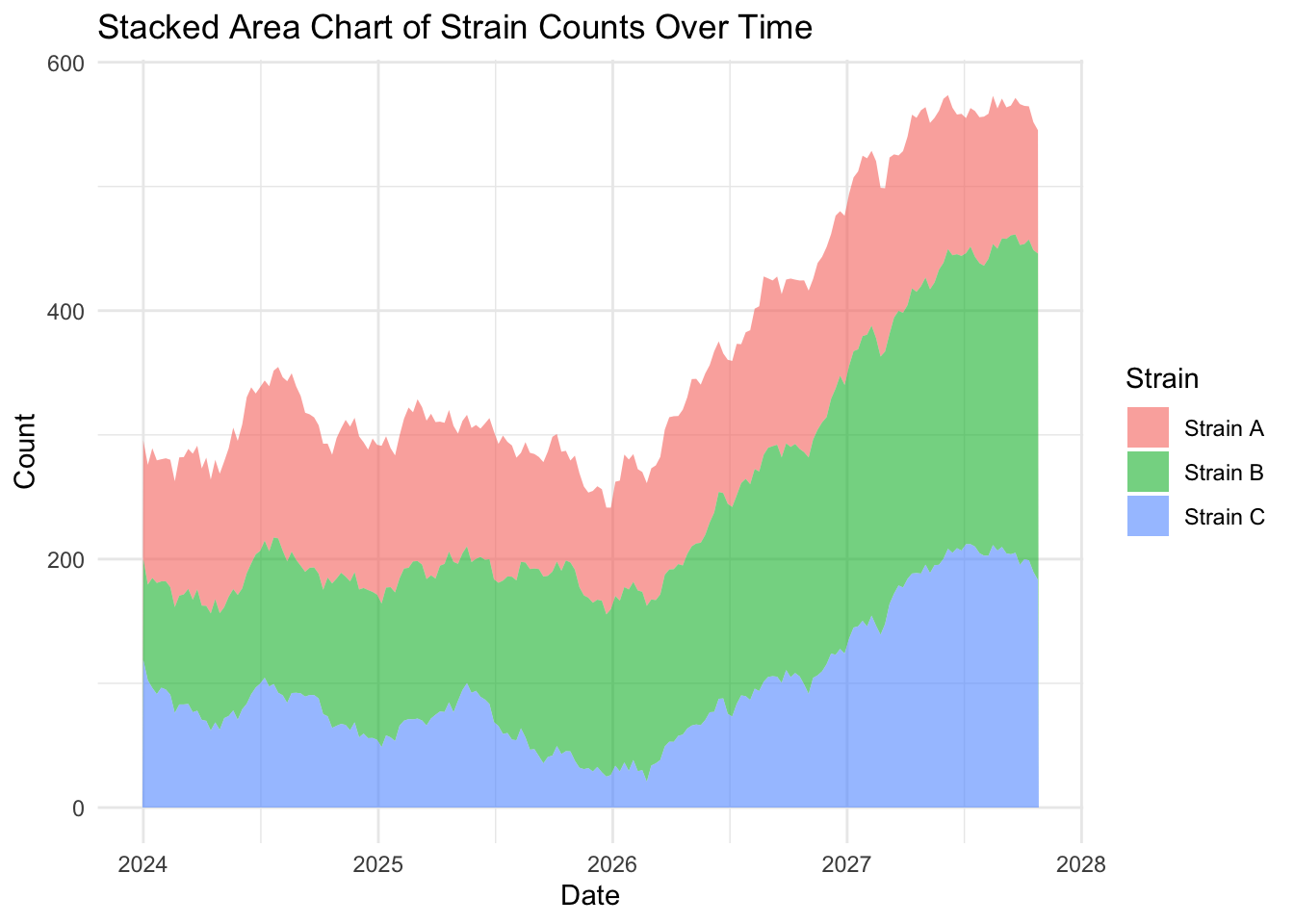
The Y-axis defaults to the count for each species. Looks like a pretty even 50/50 female/male split in the data, despite large differences in the overall individual observations for each species.
We can use a few different arguments to customise the format. The default argument used in geom_bar() is position="stack" which is would return the same plot as above. Change the geom_bar argument to position="fill" to scale our plot to percentages rather than count. The scale_y_continuous(labels = scales::percent) argument converts the y-axis labels to percentages:
ggplot(penData, aes(x = species, fill = sex)) +
geom_bar(position="fill") +
scale_y_continuous(labels = scales::percent) +
theme_classic()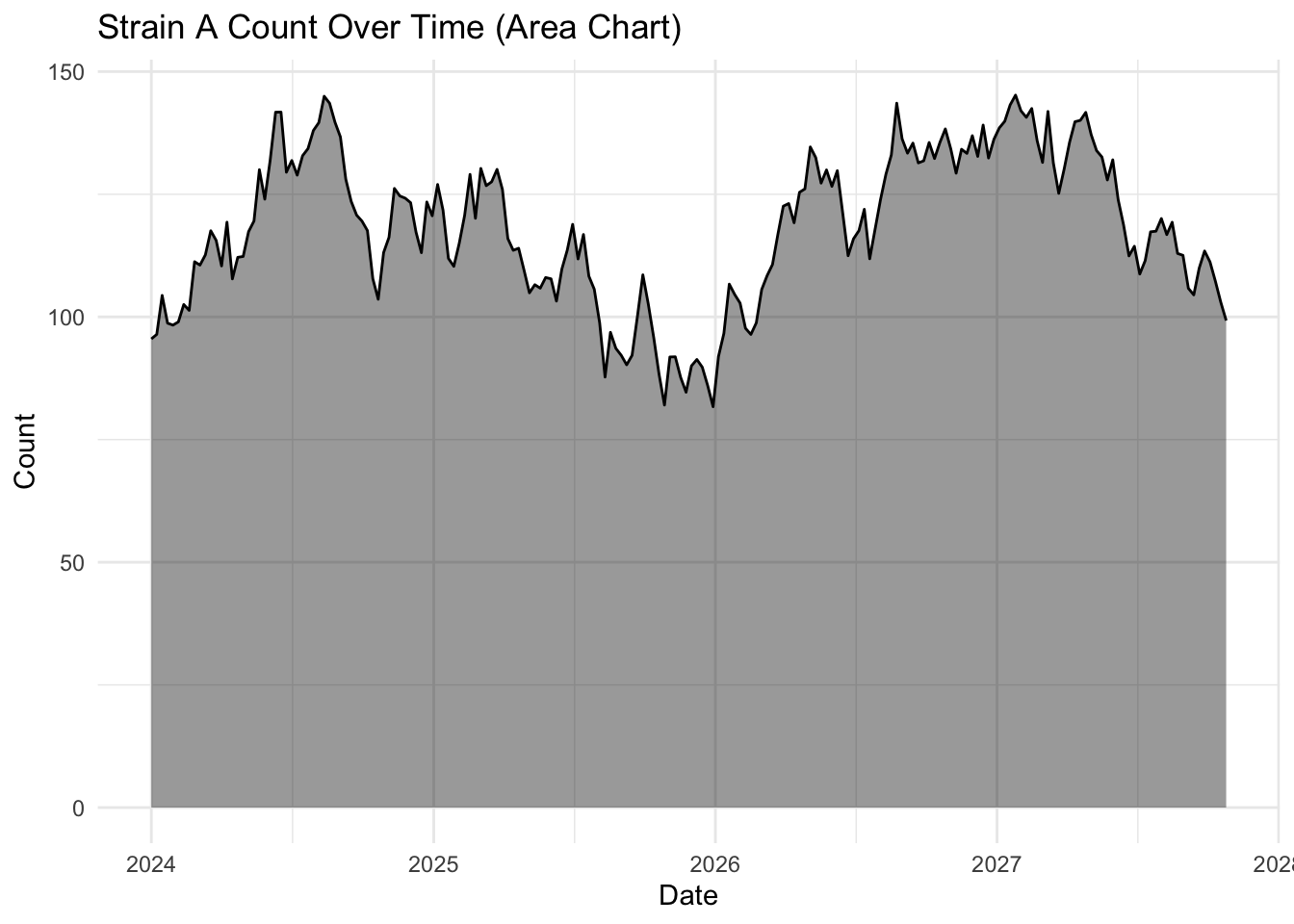
This is an opportunity to introduce you to the excellent resource that is the R-graph-gallery. We will scroll down to the “Parts of a whole” section, and walk through how this resource not only shows you different types of plots you can create, but provides you with code and reproducible examples.
You can jump straight to our first example, the waffle chart, here. The R-graph-gallery gives an overview of the type of chart and highlights some of the key ways to get started. In the case of the waffle chart we can either use a dedicated package or explore how to build a waffle chart with ggplot2 syntax. Finally, there are three examples of high quality waffle charts - click on any of these three charts to get a detailed explanation of the code used to create the chart and an example dataset.
This example below waffle chart by Muhammad Azhar is a nice one, and they have all the code required to help you build your own version.
EXERCISE 🧠🏋️♀️ (2 mins)
Back on the main page we can see that this same level of information exists for other chart types too. Take two minutes to pick one of the Parts of a Whole plots and explore some of the information about this type of chart.
Evolution (over time)
There are multiple ‘Evolution’ style plots, such as line plots, area, stacked area and time series plots. Here we will look at three variations of the line plot, which is used to show changes in a variable over time.
Generate a dataset
set.seed(12345) # Set the seed and run as one code block
# Create yeast count data for three different strains
dates <- seq.Date(from = as.Date("2026-01-01"),
to = as.Date("2026-01-31"),
by = "day")
strains <- tibble(
date = rep(dates, times = 3),
strain = rep(c("Strain A", "Strain B", "Strain C"), each = length(dates)),
count = c(
100 + cumsum(rnorm(length(dates), mean = 0, sd = 5)), # Strain A fluctuates around 100
80 + cumsum(rnorm(length(dates), mean = 0, sd = 4)), # Strain B fluctuates around 80
120 + cumsum(rnorm(length(dates), mean = 0, sd = 6)) # Strain C fluctuates around 120
)
)
What is set.seed?
What is set.seed? set.seed() is a function that feeds into any function that will randomly select or generate values. If we all use set.seed with the same number value, we will all get the same results, even though we are about to ‘randomly’ simulate some data. In future you can use any numeric value for set.seed(), so long as you keep it consistent within experiments.
Line chart with geom_line()
The simplest plot for change over time is geom_line(). The connected scatterplot is almost the same plot, but each observation is represented by a point.
ggplot(strains |> filter(strain == "Strain A"),
aes(x = date,
y = count)) +
geom_line() +
labs(title = "Strain A Count Over Time",
x = "Date",
y = "Count") +
theme_minimal()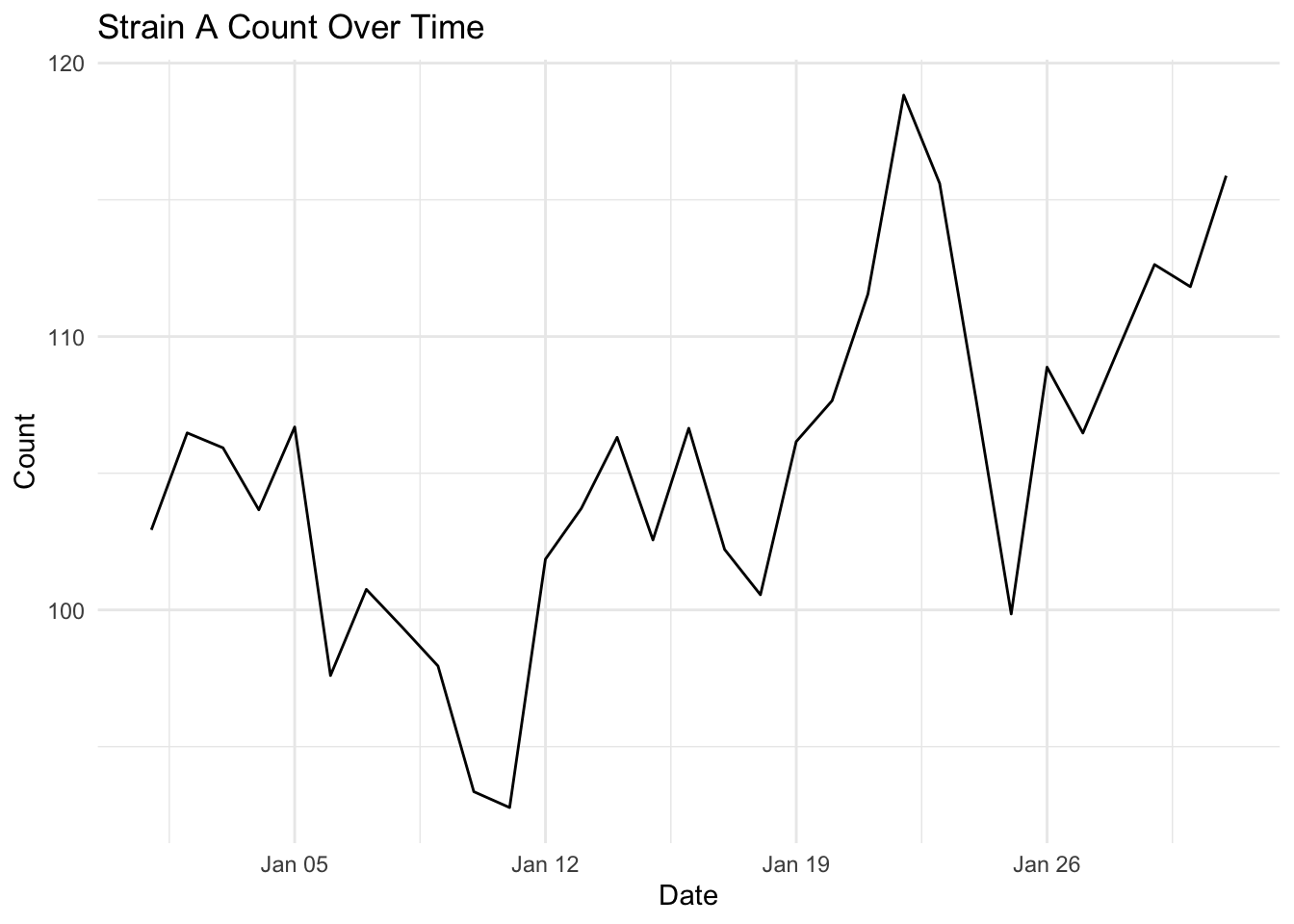
Visual interest with geom_area()
The geom_area() function uses the same principal but also includes a filled or shaded area below the line. This is the same data, but due to the shading makes the whole plot look less empty.
ggplot(strains |> filter(strain == "Strain A"),
aes(x = date, y = count)) +
geom_area(fill = "black",
alpha = 0.4) +
geom_line(color = "black",
linewidth = 0.5) + # Keeps the line for clarity
labs(title = "Strain A Count Over Time (Area Chart)",
x = "Date",
y = "Count") +
theme_minimal()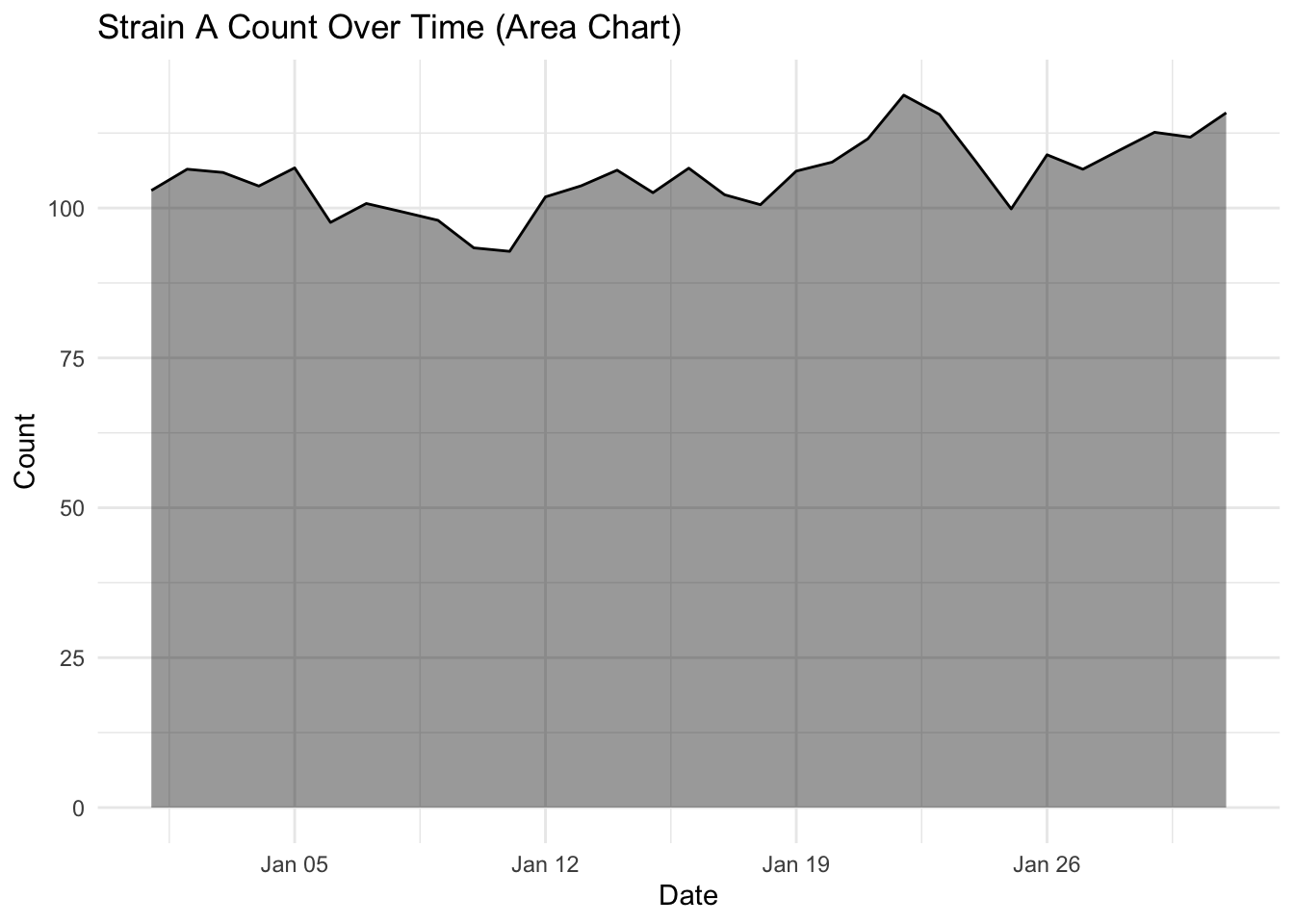
The choice of geom_line() or geom_area() is almost purely aesthetic, but you will notice the topology of these plots look a little different, even though they are both for Strain A.
DISCUSSION 🤔 (2 mins)
Why does the topology of the strain A geom_line() and geom_area() look so different?
SOLUTION
geom_line() has no baseline, so the y-axis autoscales to the range of the data, making the fluctuations look much more dramatic.
geom_area() anchors to the baseline, visually compressing the fluctuation.
To make these plots more similar, you could choose to anchor the charts using custom y-axis limits with the coord_cartesian() function.
Example 1: Anchor the y-axis to the minimum and maximum counts in your counts dataframe, to maintain consistency across all plots:
ggplot(strains |> filter(strain == "Strain A"),
aes(x = date, y = count)) +
geom_area(fill = "black", alpha = 0.4) +
geom_line(color = "black", linewidth = 0.5) +
coord_cartesian(ylim = c(min(strains$count), max(strains$count))) +
labs(title = "Strain A Count Over Time (Area Chart)", x = "Date", y = "Count") +
theme_minimal()
Example 2: Anchor the y-axis to custom coordinates to keep the fluctuation more visible:
ggplot(strains |> filter(strain == "Strain A"),
aes(x = date, y = count)) +
geom_area(fill = "black", alpha = 0.4) +
geom_line(color = "black", linewidth = 0.5) +
coord_cartesian(ylim = c(75, 120)) +
labs(title = "Strain A Count Over Time (Area Chart)", x = "Date", y = "Count") +
theme_minimal()
Stacked area chart
The stacked area chart can be used to create a strong visual impact, especially if the correct colours are chosen. Here’s an awesome example from Cédric Scherer:
Let’s create a stacked area charts of our strains, by adding the argument fill = strain to our global mapping parameters:
ggplot(strains, aes(x = date,
y = count,
fill = strain)) +
geom_area(alpha = 0.6) + # Fill areas with transparency
labs(title = "Stacked Area Chart of Strain Counts Over Time",
x = "Date",
y = "Count",
fill = "Strain") +
theme_minimal()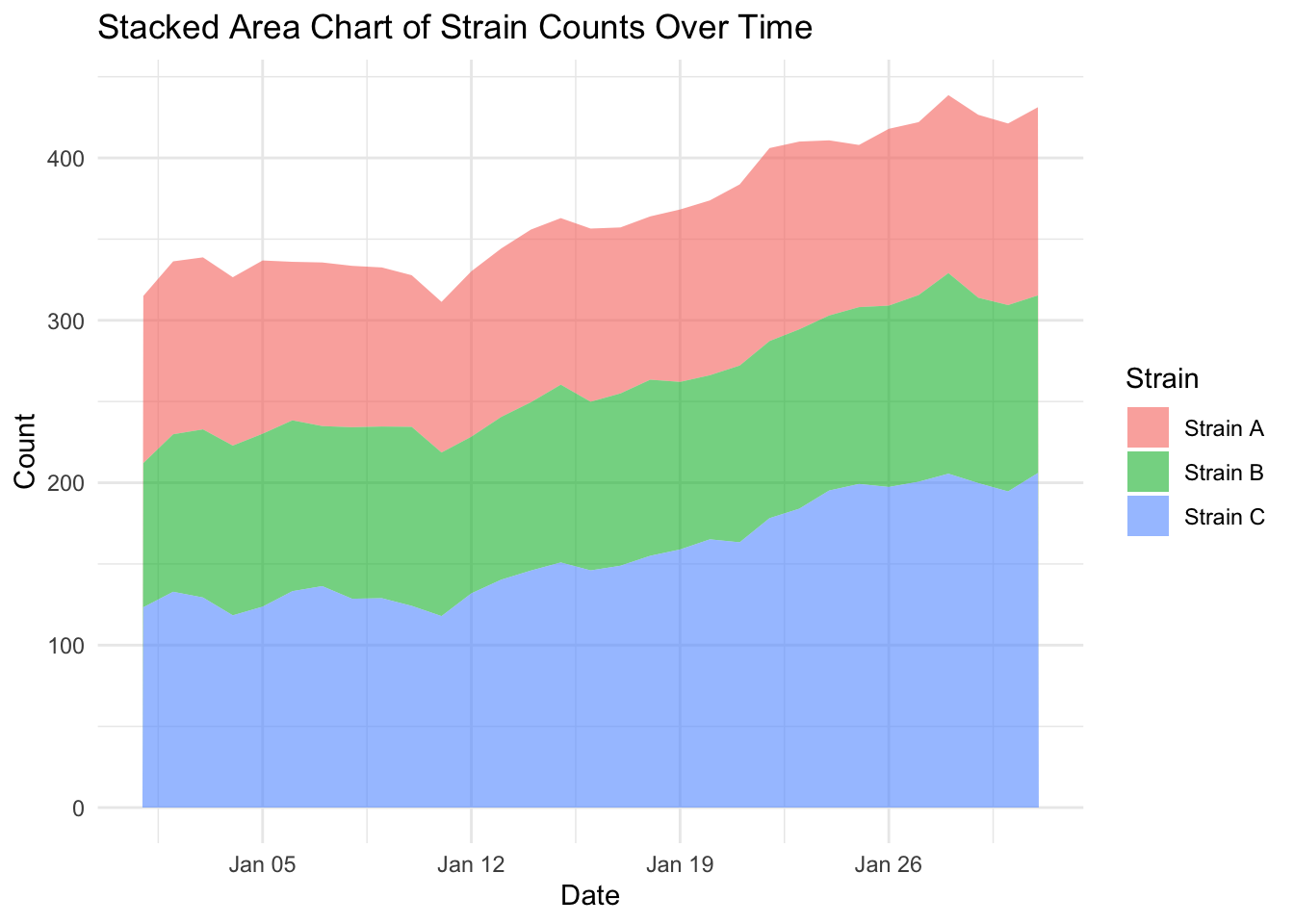
EXERCISE 🧠🏋️♀️ (4 mins) On the dubious rise of our strains…
…and the flaw with stacked area charts
Looking at the stacked area plot you would be forgiven for thinking all three strains have increased over time. Because stacked area plots can be misleading, they are not always recommended as a type of visualisation.
Investigate more closely each strain count, by changing one strain at a time filter(strain == "Strain A") (and updating the figure title of course) and plotting the area and line chart. What do you notice about the fluctuation over time for each strain? Is this visualised well in our stacked area chart (where fill = strain)?
SOLUTION
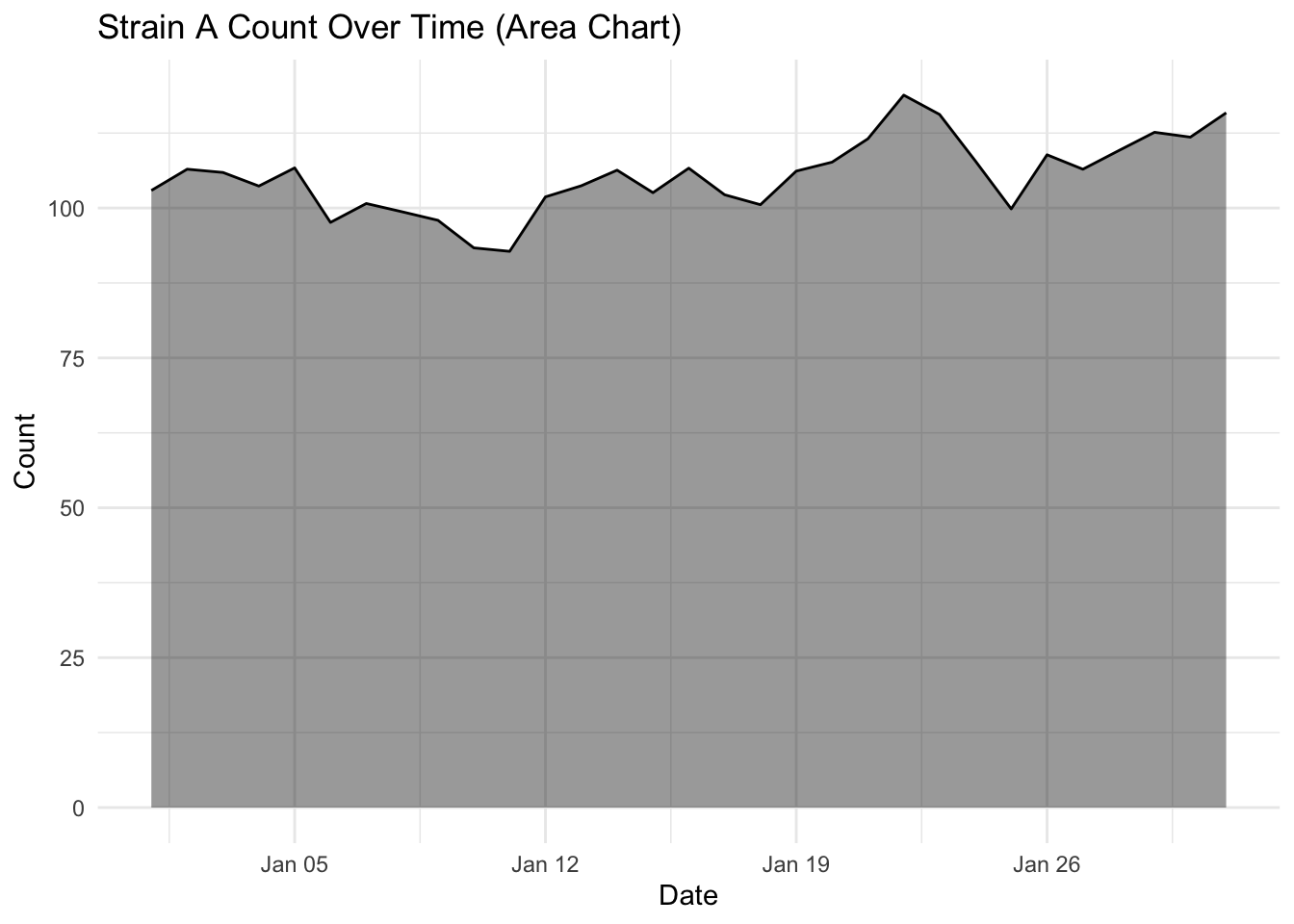
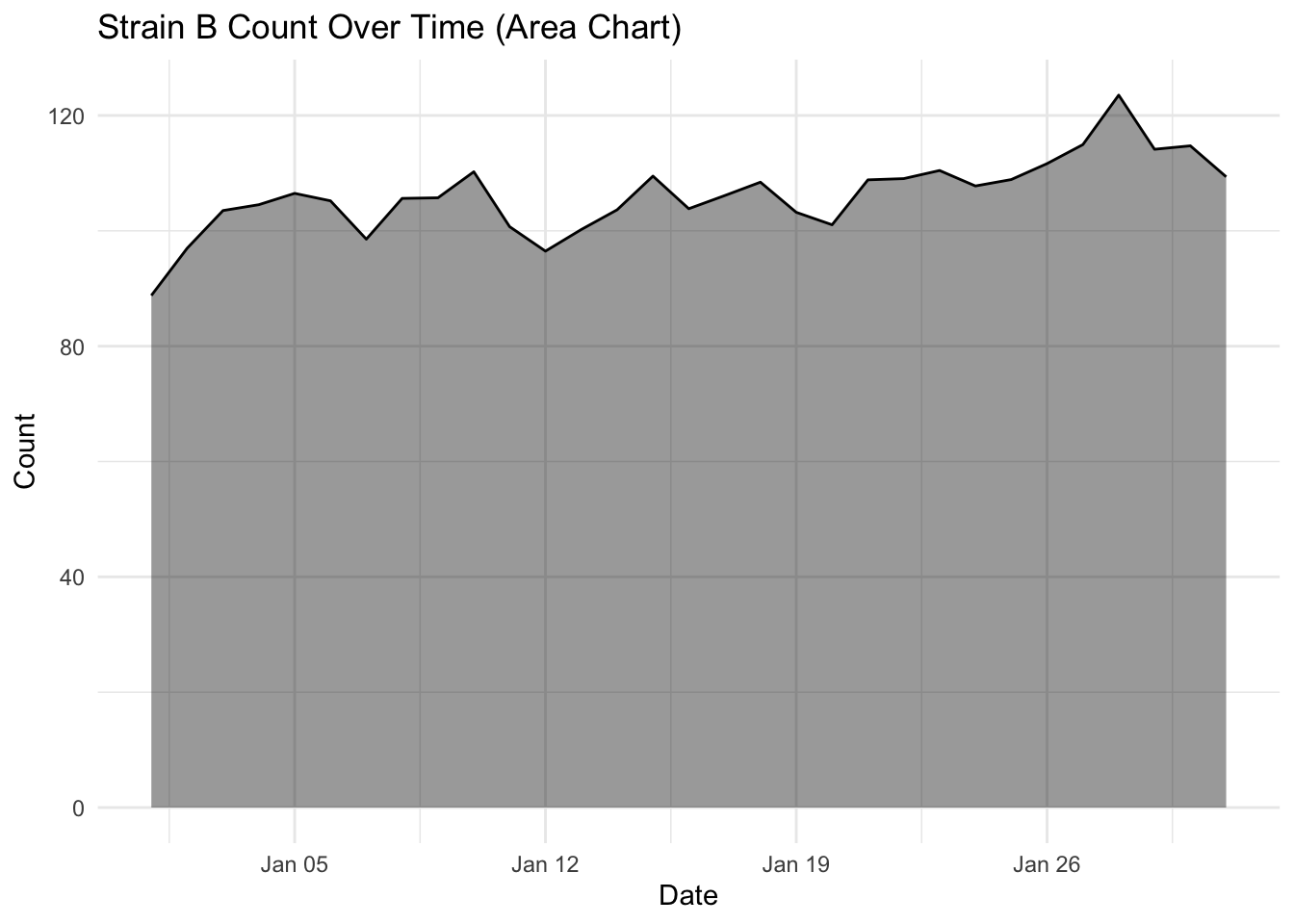
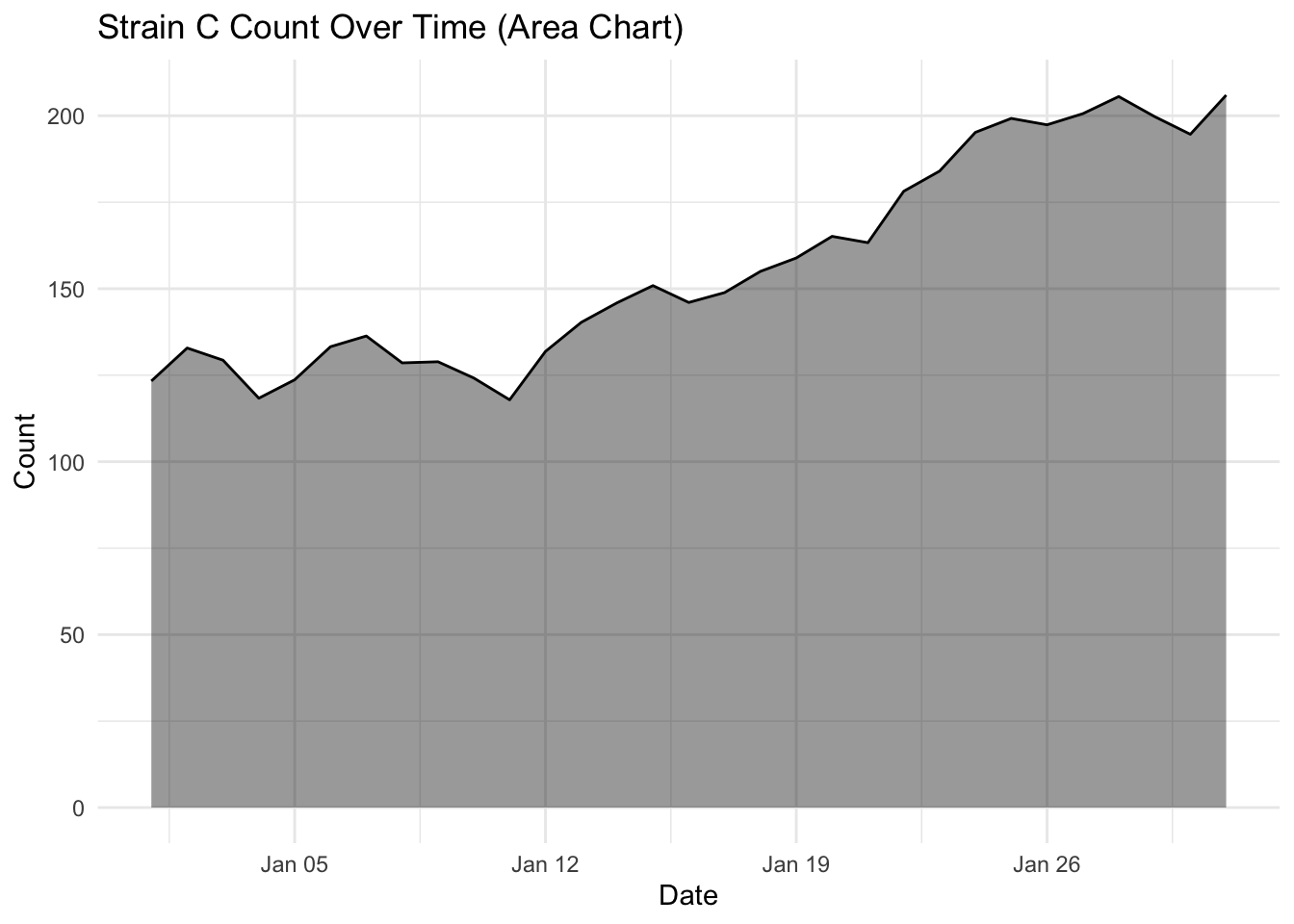
What you will notice that Strain A and B have increased in count a little over time, where as Strain C goes up quite a bit. But in our stacked area chart, it appears that every strain goes up uniformally.
You should only use a stacked area chart when you are interested in the total value of all variables together, and not the individual values of each variable (e.g., parts of a whole, percentages summing to 100%).
An alternative to stacked area plots is the use of facet, which will allow us to visualise multiple strains at once by creating individual panels (e.g., try add + facet_grid(strain ~ .) to the stacked area code block earlier. Swapping the tilde, dot and stock order (. ~ strain) rearranges the layout to vertical instead of horizontal).
Summary
The two key messages from this episode are:
There are lots of different chart types available to you, and they mostly follow the same template with slight variations in the
geom_*()function and the arguments supplied.How you visualise your data can have an impact on what other people see. The human brain is amazing, but even on our best days we make assumptions, or can be distracted, and reach the wrong conclusion. Strive to make your data as clear and transparent as possible.