library(tidyverse)
top_group_counts <- read.delim("https://raw.githubusercontent.com/GenomicsAotearoa/visualization_day/refs/heads/main/data/eDNA_group_counts.tsv")Building Better Plots
The purpose of this episode is to:
- solidify the template of ggplot2 in your mind so that you can remember and confidently create a basic plot
- introduce new geoms, familiarise ourselves with the use of arguments for different visual looks
This episode follows the outline produced by the very skilled data visualiser Cédric Scherer, modified to work with some example data and updated with my own thoughts and opinions. Cédric’s original tutorial is well worth looking at, as they are a real expert in the field of visualisation.
The data
The example dataset we are using for this session comes from Wilderlab, a company based in Aotearoa who provide eDNA monitoring and testing services (primarily from water samples). When a client orders DNA testing through Wilderlab they have the option of making their results public as a way to “provide a useful resource for scientists, conservationists, educators, and anyone else with an interest in Aotearoa’s biodiversity, water quality and biosecurity”. We will access a small subset of the data which I have accessed and curated for the workshop. It is currently stored on github as a .tsv file.
eDNA sequences are assigned a taxonomy, and when multiple sequences map to the same taxonomic unit (in our case to the level of the same genus), those additional sequences contribute to the variable called Count. I selected the 10 Groups (a classification level, e.g., Birds, Fish, Diatoms, Plants) with the highest number of sequences (that is, the groups with the most species diversity), randomly selected 500 of these sequences for each group, and saved the data to a file. We can now look at Count (the abundance) of these sequences and see if this varies across Groups.
A contrived example
For the sake of honesty, if the above explanation of how I created our data subset sounds arbitrary - it is. Originally I wanted an example look at the relationship between Group diversity (number of species) and Count (abundance). I hypothesised that a Group like mammals would have low diversity but high counts, while a Group like Insects would have high diversity but lower counts (based on what I know about animal diversity and how I think body size will impact eDNA abundance). Notably, some Groups had a lot of diversity, and as I built the workshop and some of the figures I realised that having too many sequences made the data points difficult to differentiate. Eventually I trimmed the original data set so that each group had 500 randomly selected sequences. This made visualisation easier, but forced me to re-work the example.
Familiarise yourself with the data
Here we will use some functions to check features of our data
top_group_counts |> head() Name CommonName TaxID Count
1 Anas platyrhynchos Mallard duck 8839 273
2 Pavo cristatus Indian peafowl; common peafowl; blue peafowl 9049 33
3 Carduelis carduelis Goldfinch 37600 6
4 Porphyrio melanotus Pukeko 72013 1361
5 Carduelis carduelis Goldfinch 37600 20
6 Zosterops White eye 36298 13
Rank Group Phylum Class Order Family Genus
1 species Birds Chordata Aves Anseriformes Anatidae Anas
2 species Birds Chordata Aves Galliformes Phasianidae Pavo
3 species Birds Chordata Aves Passeriformes Fringillidae Carduelis
4 species Birds Chordata Aves Gruiformes Rallidae Porphyrio
5 species Birds Chordata Aves Passeriformes Fringillidae Carduelis
6 genus Birds Chordata Aves Passeriformes Zosteropidae Zosteropstop_group_counts |> tail() Name CommonName TaxID Count Rank Group
4995 Nais Sludgeworm 74730 101 genus Worms
4996 Dero Worm 66487 72 genus Worms
4997 Cernosvitoviella aggtelekiensis Worm 913639 13 species Worms
4998 Octolasion lacteum Worm 334871 36 species Worms
4999 Lumbriculus variegatus Blackworm 61662 3848 species Worms
5000 Limnodrilus hoffmeisteri Redworm 76587 19 species Worms
Phylum Class Order Family Genus
4995 Annelida Clitellata Tubificida Naididae Nais
4996 Annelida Clitellata Tubificida Naididae Dero
4997 Annelida Clitellata Enchytraeida Enchytraeidae Cernosvitoviella
4998 Annelida Clitellata Crassiclitellata Lumbricidae Octolasion
4999 Annelida Clitellata Lumbriculida Lumbriculidae Lumbriculus
5000 Annelida Clitellata Tubificida Naididae Limnodrilustop_group_counts |> class()[1] "data.frame"# Verify that all groups have 500 observations
top_group_counts |>
group_by(Group) |>
summarize(observations = n(), .groups = "drop") |>
arrange(desc(observations)) |>
head()# A tibble: 6 × 2
Group observations
<chr> <int>
1 Birds 500
2 Ciliates 500
3 Diatoms 500
4 Fish 500
5 Insects 500
6 Mammals 500Initial visualisation
In this example we are looking at the variance in Count across the 10 groups. A very reasonable starting point for this type of visualisation is the boxplot. We will use a basic boxplot to get a ‘sketch’ of what our visualisation will look like, then we will test other geoms (plot types) and look at colours, titles, labels etc.,.
Boxplot 101
A basic boxplot:
ggplot(data = top_group_counts,
mapping = aes(x = Group, y = Count)) +
geom_boxplot()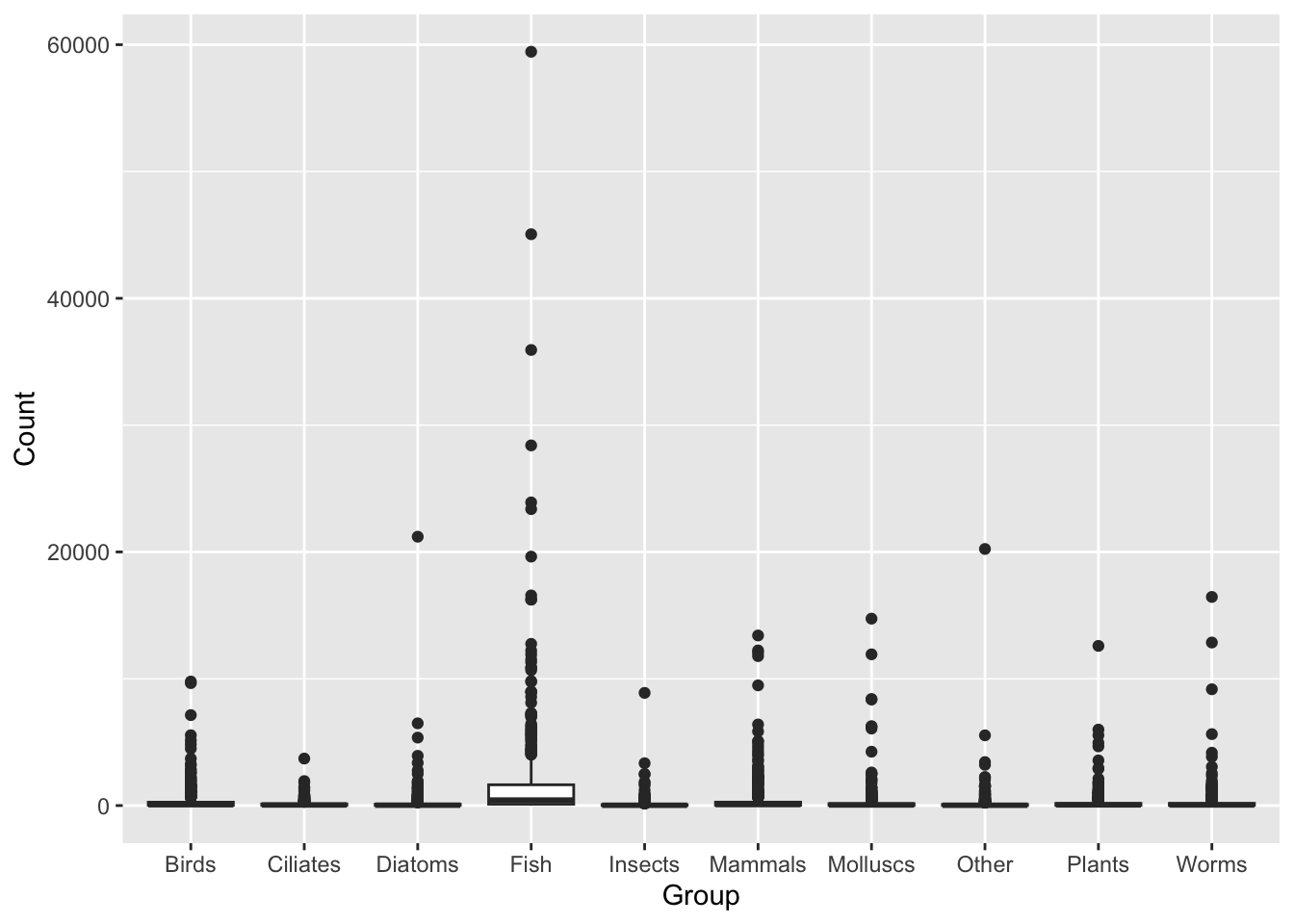
This figure shows us the Count variable is highly skewed (the majority of values are small, with some values many fold higher).
EXERCISE 🧠🏋️♀️ (3 mins)
Use the summary() function on the top_group_counts dataset and look at the outcome for the Count variable. What does this tell us about how we need to plot the data?
More advanced R users could use the filter() and nrow() functions to ask how many rows have a Count value that is 10 times greater than the Count median (or ask this question using whatever functions you are familiar with).
Solution
top_group_counts |> summary() Name CommonName TaxID Count
Length :5000 Length :5000 Min. : 2706 Min. : 5.0
N.unique : 871 N.unique : 425 1st Qu.: 9940 1st Qu.: 17.0
N.blank : 0 N.blank :1005 Median : 76587 Median : 51.0
Min.nchar: 3 Min.nchar: 0 Mean : 401972 Mean : 421.9
Max.nchar: 43 Max.nchar: 81 3rd Qu.: 238745 3rd Qu.: 197.2
Max. :10000288 Max. :59445.0
Rank Group Phylum Class
Length :5000 Length :5000 Length :5000 Length :5000
N.unique : 5 N.unique : 10 N.unique : 17 N.unique : 44
N.blank : 0 N.blank : 0 N.blank : 0 N.blank : 0
Min.nchar: 5 Min.nchar: 4 Min.nchar: 8 Min.nchar: 4
Max.nchar: 10 Max.nchar: 8 Max.nchar: 16 Max.nchar: 19
NAs : 45 NAs : 178
Order Family Genus
Length :5000 Length :5000 Length :5000
N.unique : 184 N.unique : 365 N.unique : 620
N.blank : 0 N.blank : 0 N.blank : 0
Min.nchar: 6 Min.nchar: 6 Min.nchar: 3
Max.nchar: 18 Max.nchar: 20 Max.nchar: 22
NAs : 189 NAs : 105 Note the outputs, specifically that for Count. Min = 5.0, Median = 51.0, Mean = 421.9, Max = 59445. This is a sign the data is skewed. We will need to take this into account when we plot the data, otherwise the majority of our data points will be squished together in a small area of the plot, and we won’t be able to see any differences between groups. These data will need to be transformed using a log transformation.
Another way we could investigate this data is by calculating how many rows have a Count value that is more than 10 times greater than the median:
top_group_counts |>
filter(Count >= median(top_group_counts$Count)*10) |>
nrow()[1] 681This shows 681 elements are more than 10 times higher than the median - very skewed.
Transform the data for visualisation
We can address the issue of skewed data by taking the log of Count before we plot it.
ggplot(data = top_group_counts,
mapping = aes(x = Group, y = log(Count))) +
geom_boxplot()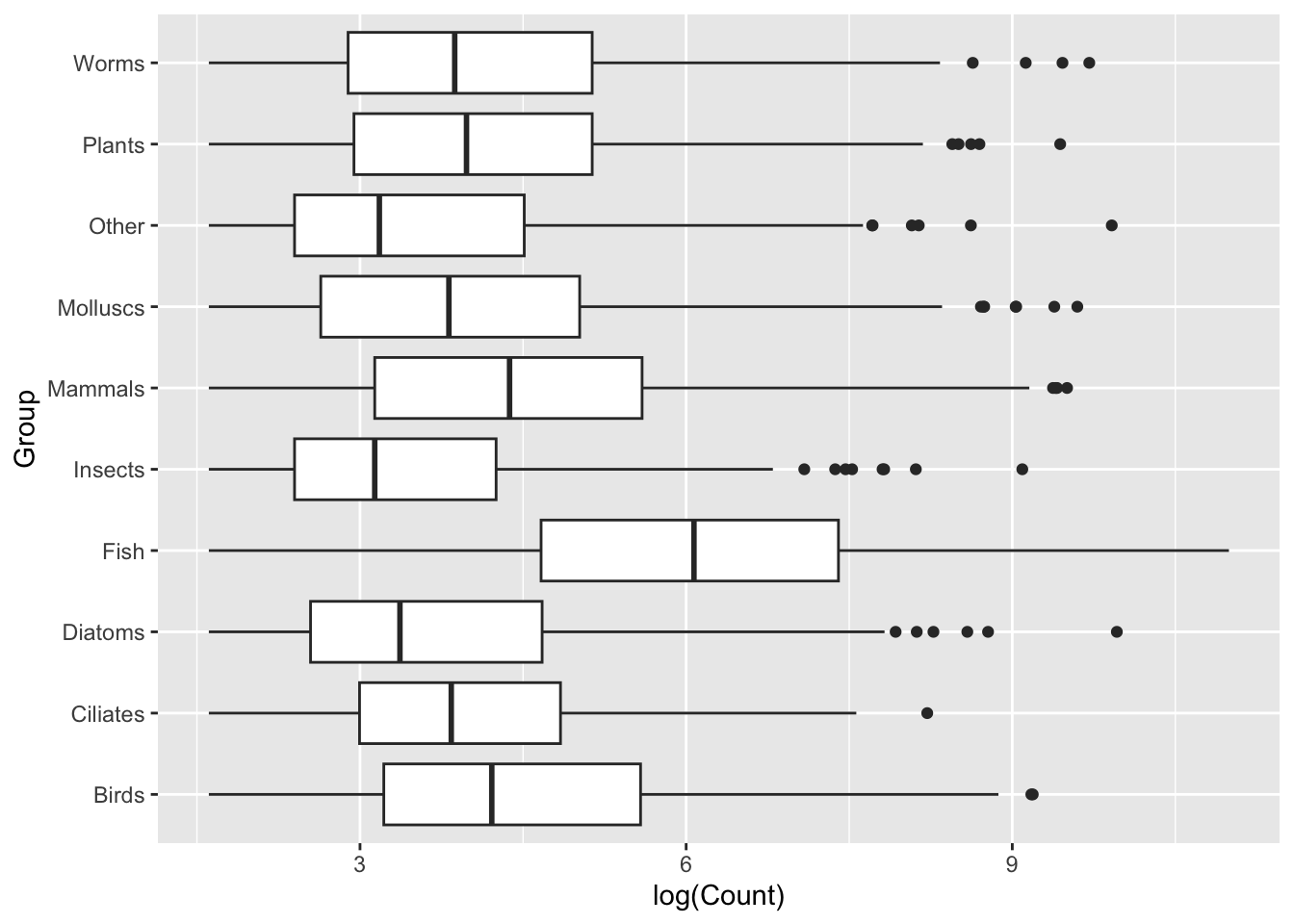
Design tip: It can often be worth representing boxplots on a horizontal, rather than vertical, distribution. Let’s switch our axes assignments:
ggplot(data = top_group_counts,
mapping = aes(x = log(Count), y = Group)) +
geom_boxplot()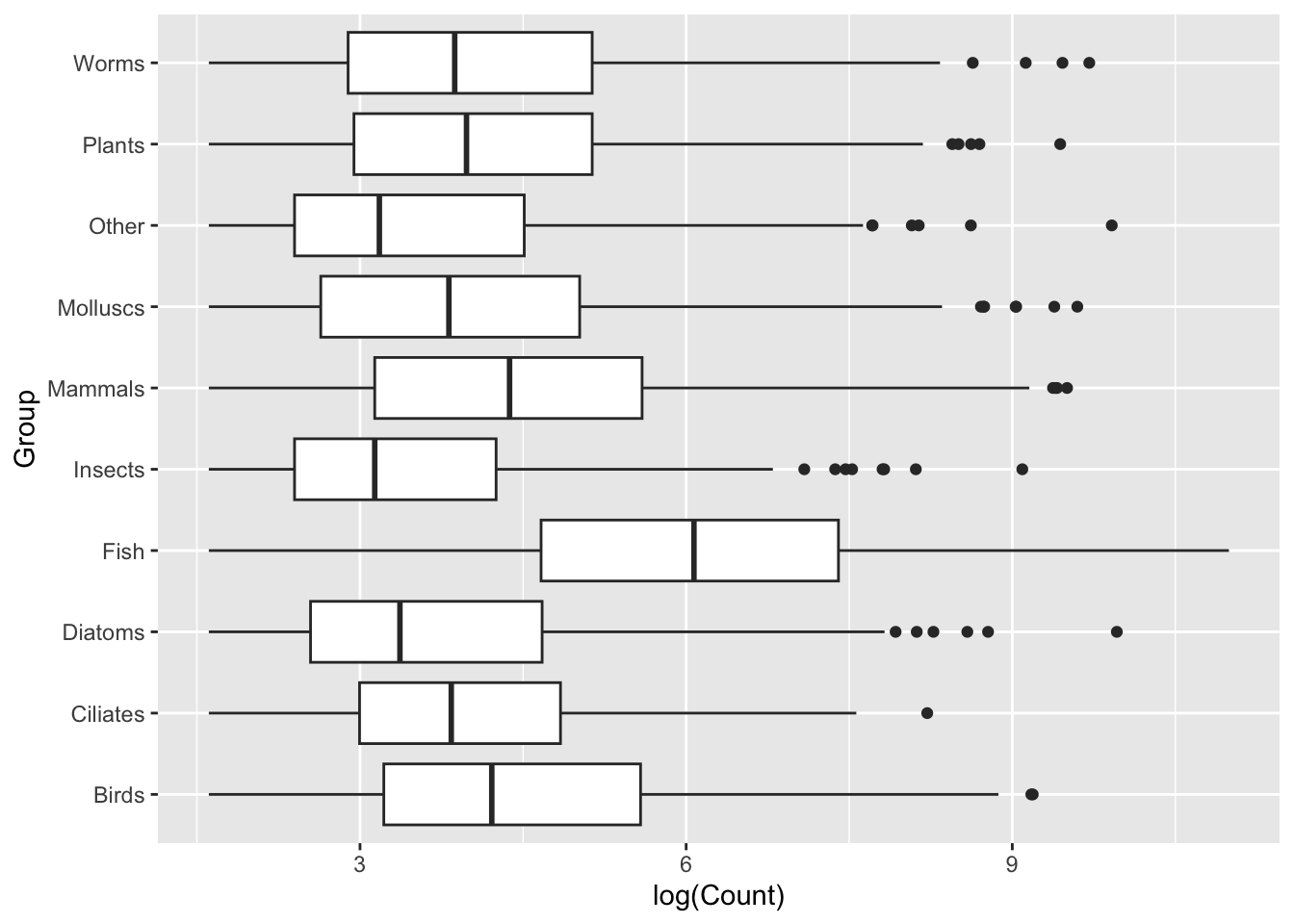
Generally we will find that sometimes switching the layout like this can provide us with clearer visualisation, sometimes due to labels, sometimes for aesthetics. In this case, I think the group labels read much more clearly when stacked vertically on top of one another with the new layout. I encourage you to think about small changes like this when creating your visualisations.
Sort the data
Sorted data will be easier to interpret, with a lower cognitive load. Note: we should only sort data if there is no inherent structure in the categories. Mammals are not inherently ‘higher’ than Fish, so these groups can be sorted based on numerical summary of the data. Sometimes we will have internal structure, either some kind of hierarchy or grouping (e.g., treatment vs control, timepoints) and it is not appropriate to apply sorting.
How are we sorting?
Each group contains multiple Count values (because we are plotting boxplots). So we need a single summary statistic per group to decide the order.
Here, we use the median count for each group.
We then reorder the factor levels of Group so that groups appear from lowest median count to highest median count in the plot. This makes it visually clear which groups tend to have larger values.
We use:
fct_reorder()from forcats to reorder factor levelsmedianas the summary statisticmutate()to update the existingGroupvariable
top_group_counts_sorted <- top_group_counts |>
mutate(Group = fct_reorder(Group, Count, .fun = median))What this does: For each Group, calculate the median of Count, then reorder the levels of Group based on those median values.
Use
factor() to factorise the Group column instead:
The fct_reorder() function comes from the forcats package from tidyverse, but base R has its own factorising function factor(). You may be more familiar with how factor() works, so let’s look at how we could achieve the above with that (it’s a bit more convoluted!)
First we need to create a vector of the variables in Group, in the order we want them to plot in.
group_order <- top_group_counts |>
group_by(Group) |>
summarise(median_count = median(Count)) |>
arrange(median_count) |>
pull(Group)
group_order [1] "Insects" "Other" "Diatoms" "Molluscs" "Ciliates" "Worms"
[7] "Plants" "Birds" "Mammals" "Fish" The code above will group all rows by the “Group” column, to then calculate the median of the “Count” rows based on “Group” (and place this value in a new column called median_count), then arrange the median_count column from lowest to highest, and pull out the Group column as a vector.
Now that we have a vector of our Group names in the order we want them in, we can factorise our dataframe column:
top_group_counts_sorted <- top_group_counts
top_group_counts_sorted$Group <- factor(top_group_counts_sorted$Group, levels = group_order)Now plot the sorted data:
ggplot(data = top_group_counts_sorted,
mapping = aes(x = log(Count), y = Group)) +
geom_boxplot()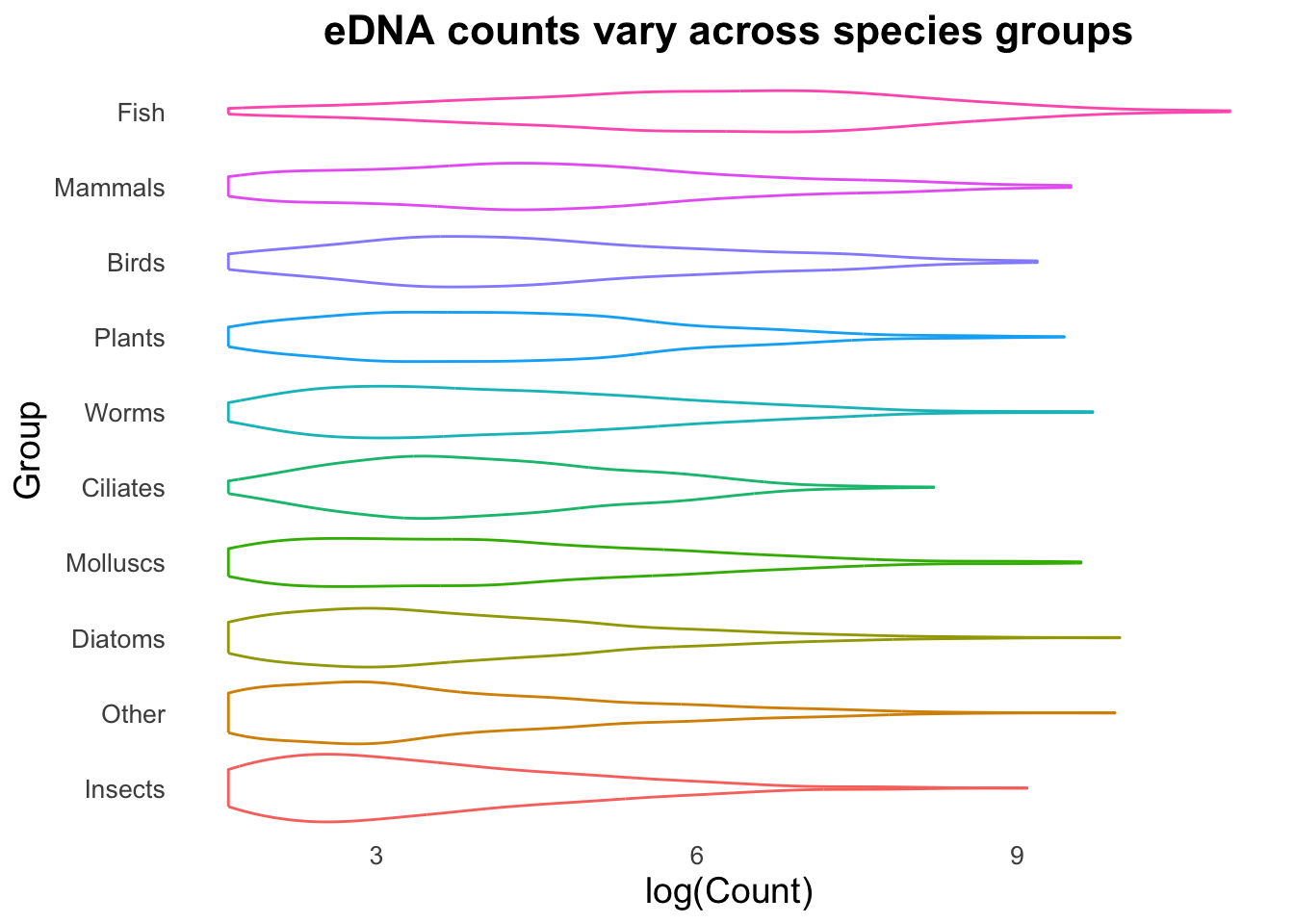
This is immediately clearer, with a considerably lower cognitive load. Easily observe that not only does the Fish group have a higher set of Counts, but it seems to break with the trend of the other Groups.
Outliers
In boxplots, outliers are identified as values that are more than 1.5 times the interquartile range above or below the third and first quartile, respectively. In our data these outliers are reasonably numerous. This could be due to inherently variable data, due either to biological or technical reasons, site differences etc.,. Without wanting to make assumptions about the data we can experiment with visualisations and choose how we want to present this data.
Better visualisation
We can now start to focus on the visualisation aspects: control plot themes such as labels, titles, spacing, group colours. We can explore geoms, and variations on geoms, to determine the best way to present the data. Finally, we can add additional geoms or mappings to display more data.
Theme is a separate function that controls almost all visual elements of a ggplot. We can fine tune text elements (font size, shape, angle for axis text, create custom labels and titles), the legend (changing the position, setting a background, control the title and content), the ‘panel’ (panel is the background of the plot, in the above cases we have a grid), and many other features.
Notice below we use a slightly different format for writing our ggplot function - we omit data = and mapping =, since those calls are always required and used.
Let’s build on the same plot we made above, but add some customisation.
ggplot(top_group_counts_sorted,
aes(x = log(Count),
y = Group,
colour = Group)) +
labs(title = "eDNA counts vary across groups",
x = "log(Count)",
y = "Group") +
theme_minimal() +
theme(
plot.title = element_text(size = 16, face = "bold", hjust = 0.5),
axis.title = element_text(size = 14),
axis.text = element_text(size = 10),
panel.grid = element_blank()
) +
geom_boxplot()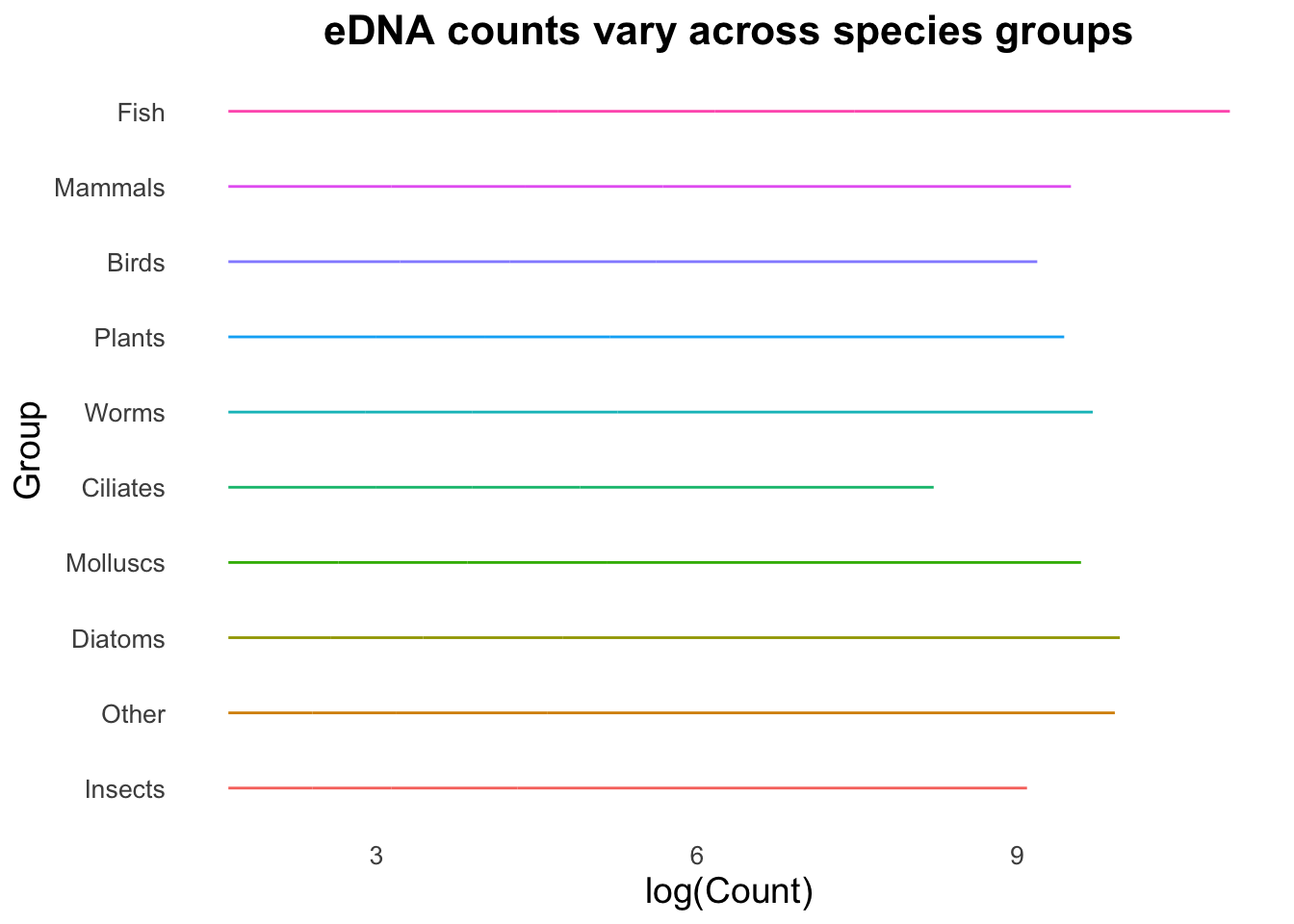
EXERCISE 🧠🏋️♀️ (10 mins)
Look at how colour has been visualised in the boxplot above. We assigned colour in aes() to each group, which has resulted in the outlines of each of the boxplots being assigned individual default R colours. Keen-eyed observers will also see we now have a legend for our Group variable. By specifying the colour or the fill in the mapping aes(), a legend will be added automatically by ggplot.
One at a time, try out these changes to the plot and note how the visualisation changes:
- Within the mapping
aes()function, changecolour = Grouptofill = Group.
- In the
theme()section,legend.position = "none"or to"left"or"right". - Add another geom layer, either
+ geom_jitter()or+ geom_point() - Change the transparency of the geoms using
alpha = 0.5and the colour or fill using theaes()function within the geom (like in point 1 with the global mappingaes()).
Pay attention too to how changes in the mapping aes() globally affect how geoms are visualised.
Now that you have had a play around with these different options, try these challenges below.
⚡️ CHALLENGE 1: Create this plot
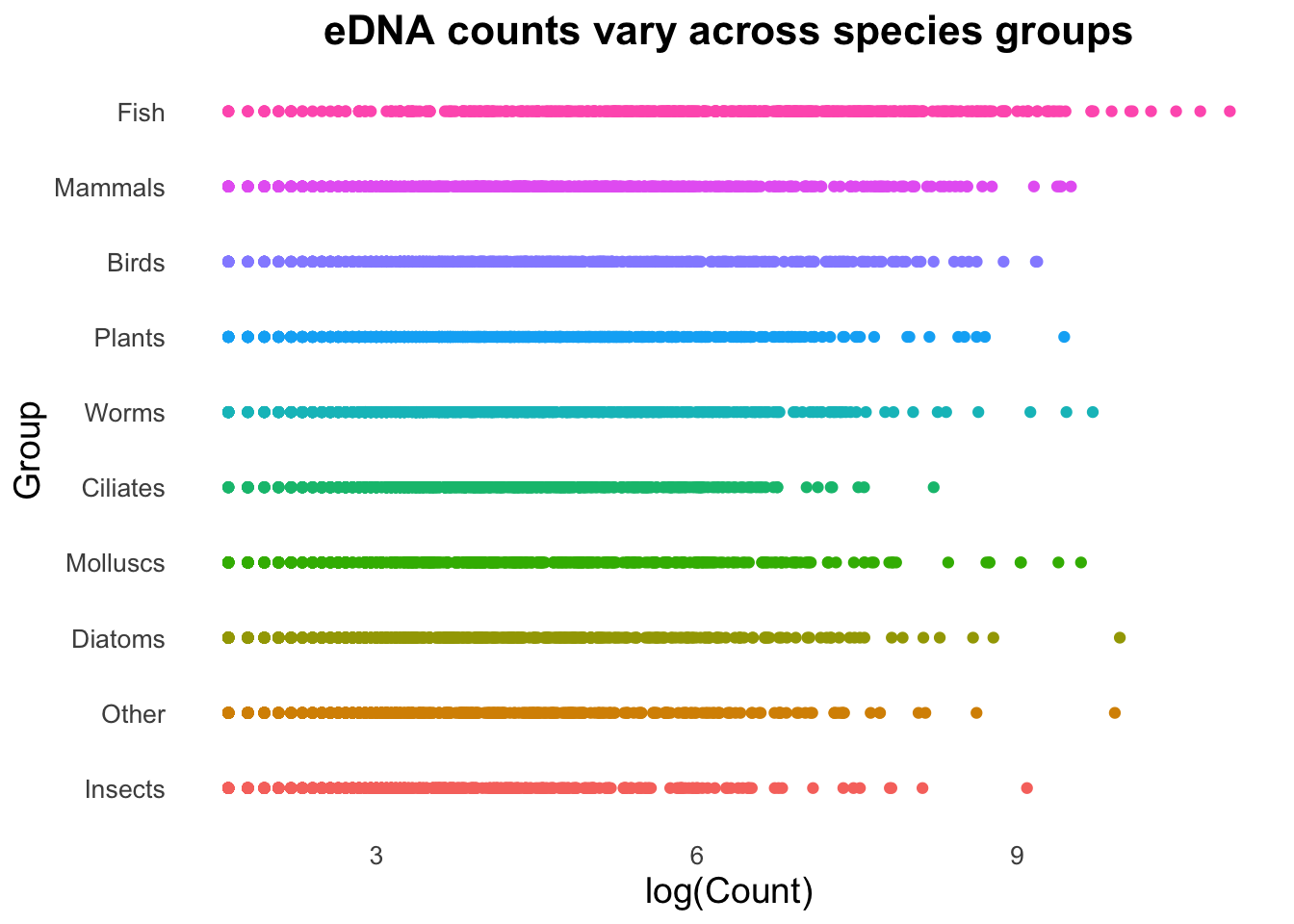
Hint: the options needed to make this plot are all in the 4 points above, where you had a play around with the different options.
SOLUTION CHALLENGE 1:
ggplot(top_group_counts_sorted,
aes(x = log(Count),
y = Group,
fill = Group)) +
labs(title = "eDNA counts vary across groups",
x = "log(Count)",
y = "Group") +
theme_minimal() +
theme(
legend.position = "none",
plot.title = element_text(size = 16, face = "bold", hjust = 0.5),
axis.title = element_text(size = 14),
axis.text = element_text(size = 10),
panel.grid = element_blank()
) +
geom_boxplot() ⚡️ CHALLENGE 2: Create this plot
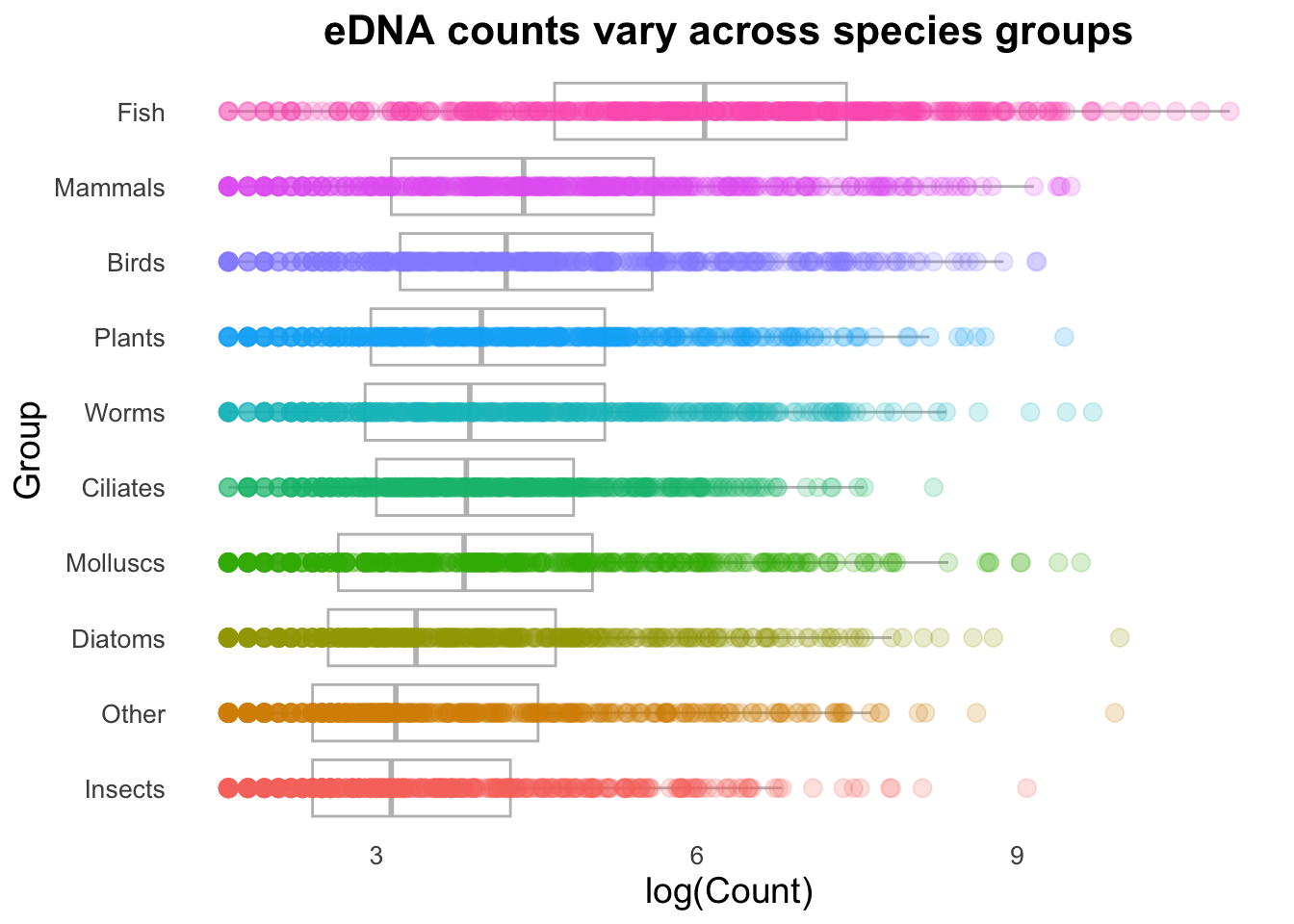
Hint: the options needed to make this plot are all in the 4 points above, where you had a play around with the different options.
SOLUTION CHALLENGE 2:
ggplot(top_group_counts_sorted,
aes(x = log(Count),
y = Group)) +
labs(title = "eDNA counts vary across groups",
x = "log(Count)",
y = "Group") +
theme_minimal() +
theme(
legend.position = "none",
plot.title = element_text(size = 16, face = "bold", hjust = 0.5),
axis.title = element_text(size = 14),
axis.text = element_text(size = 10),
panel.grid = element_blank()
) +
geom_jitter(colour="grey") +
geom_boxplot(alpha = 0.5, aes(colour = Group)) Note: the order of the layers matters! If you switch the order of the geoms, the boxplots will be drawn on top of the jitter points, and the jitter points will be obscured.
Fine tuning
A useful tip for working with ggplot is to save a set of basic features, such as the data, mapping, and theme to an object which can then be used for plotting with different geoms later.
Let’s keep the basic parameters that we like in a new object called groupCounts, without a geom, then customise from there.
groupCounts <- ggplot(top_group_counts_sorted,
aes(x = log(Count),
y = Group)) +
labs(title = "eDNA counts vary across groups",
x = "log(Count)",
y = "Group") +
theme_minimal() +
theme(
legend.position = "none",
plot.title = element_text(size = 16, face = "bold", hjust = 0.5),
axis.title = element_text(size = 14),
axis.text = element_text(size = 10),
panel.grid = element_blank()
)
Wait! Anything else you want to customise here before we move on?
You may want to further customise the text in theme() and labs() to better visualise your plot. Suggested possible changes:
- Remove the y axis title using either
labs(y = "")ortheme(axis.title.y = element_blank()) - Change the font of each label using the
family = "font of choice"argument withintheme( ... = element_text(family = "font of choice")). e.g., try “Helvetica” or “Mukta” - Change font/text size of specific axis text e.g., using
theme(axis.text.x = element_text(size = #))
Size, alpha, colour
A particularly useful argument with the geom_point() function is alpha, which controls the opacity/transparency of the points. You can think of alpha like the amount of ink being printed on a page, with 1.0 as being like 100% ink and 0.5 as 50% ink. With reduced opacity we can see more clearly when points are placed on top of one another. It is less clear in this example because there are so many data points in a small space, but it’s usefulness will become more apparent shortly. Let’s map the colour here too to our Group variable using aes(colour = Group).
groupCounts +
geom_point(size = 3,
alpha = 0.2,
aes(colour = Group))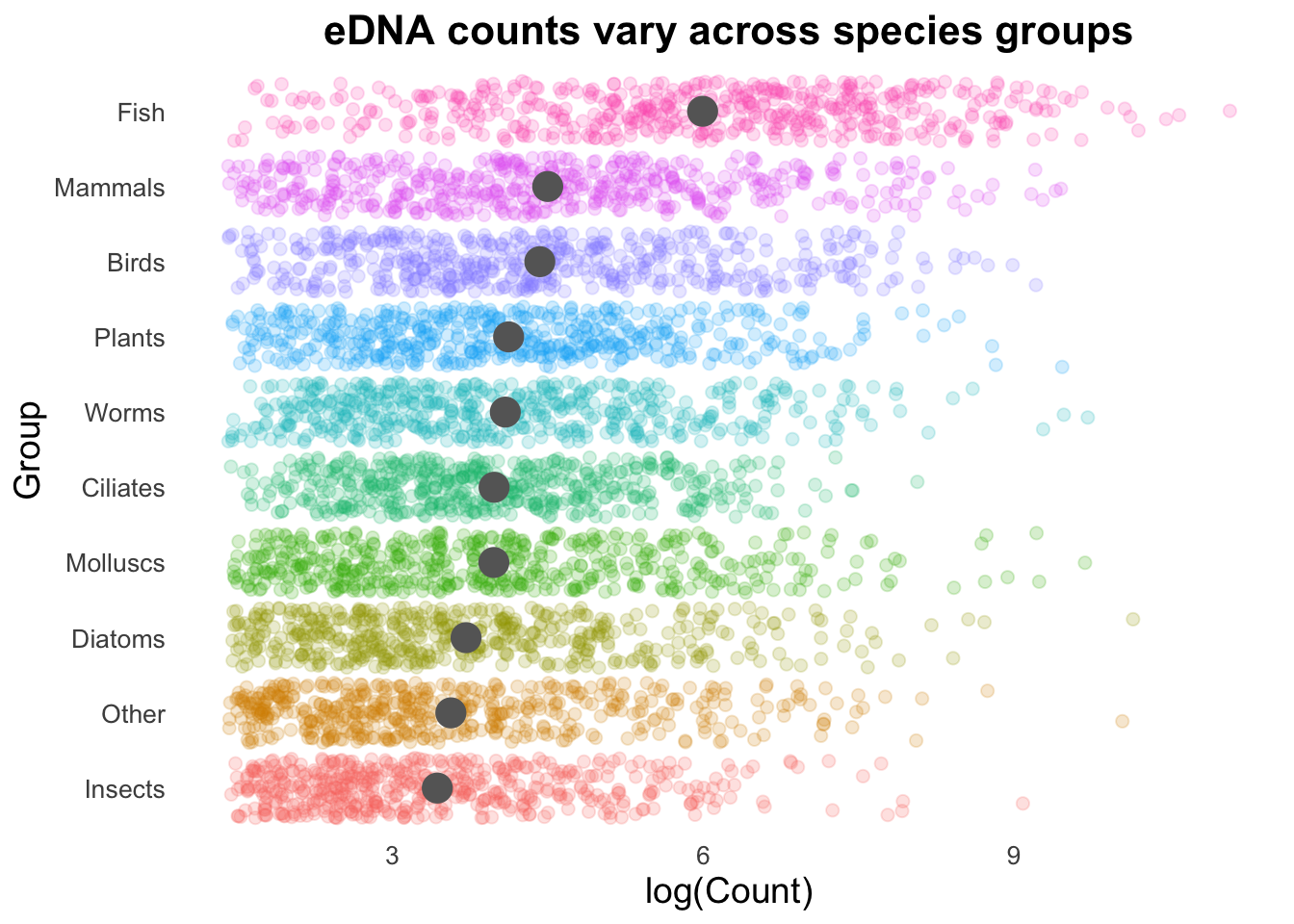
Why does colour have to be within the
aes() function here?
Try these two blocks of code and compare the difference:
groupCounts +
geom_point(size = 3,
alpha = 0.2,
colour = Group)groupCounts +
geom_point(size = 3,
alpha = 0.2,
colour = "red")
SOLUTION
In the first block of code, colour is outside of the aes() function, so it is not mapped to the Group variable. Instead, ggplot looks for an object called Group in the global environment and doesn’t find it, so it throws an error.
Output 1:
Error: object 'Group' not foundIn the second block of code, colour is set to “red”, which is a fixed value. This means that all points will be coloured red (and there will be no legend for Group – but we set it as legend = none within our groupCounts object anyway).
Output 2:
Geoms on geoms on geoms
Geoms can be combined to create more complex plots by layering one set of information over another.
groupCounts +
geom_boxplot(colour = "gray",
outlier.alpha = 0) +
geom_point(size = 3,
alpha = 0.2,
aes(colour = Group))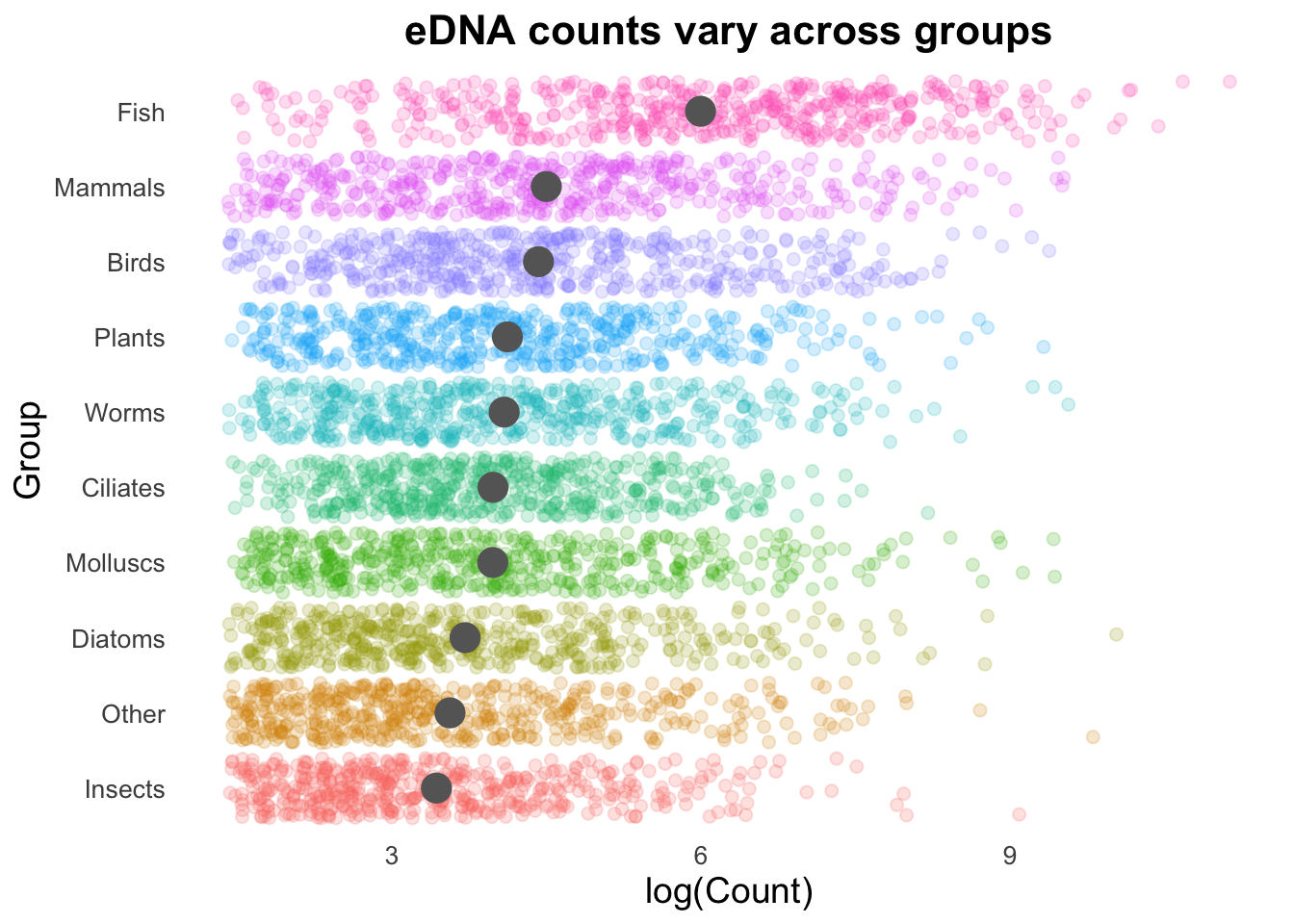
Note that we’ve included a new argument in geom_boxplot(outlier.alpha = 0). The boxplot geom normally adds points that are considered outliers (greater than 1.5 times the interquartile range above or below the 1st or 3rd quartile). Since these points are going to be represented by the geom_point() function, we need to set outlier.alpha to 0 so that they are functionally invisible (and are therefore not printed twice).
While the above demonstrates the concept of combining geoms, it’s not very functional. The high density of our points on a straight line is creating a strong effect that is over-powering other visualisations. We can change the points from being on a single line to spaced apart with the geom_jitter() function, which adds random horizontal movement, and we can use the argument width = 0.2 to control how far apart the points are spaced.
groupCounts +
geom_boxplot(colour = "gray40",
outlier.alpha = 0) +
geom_jitter(size = 2,
alpha = 0.2,
width = 0.2,
aes(colour = Group))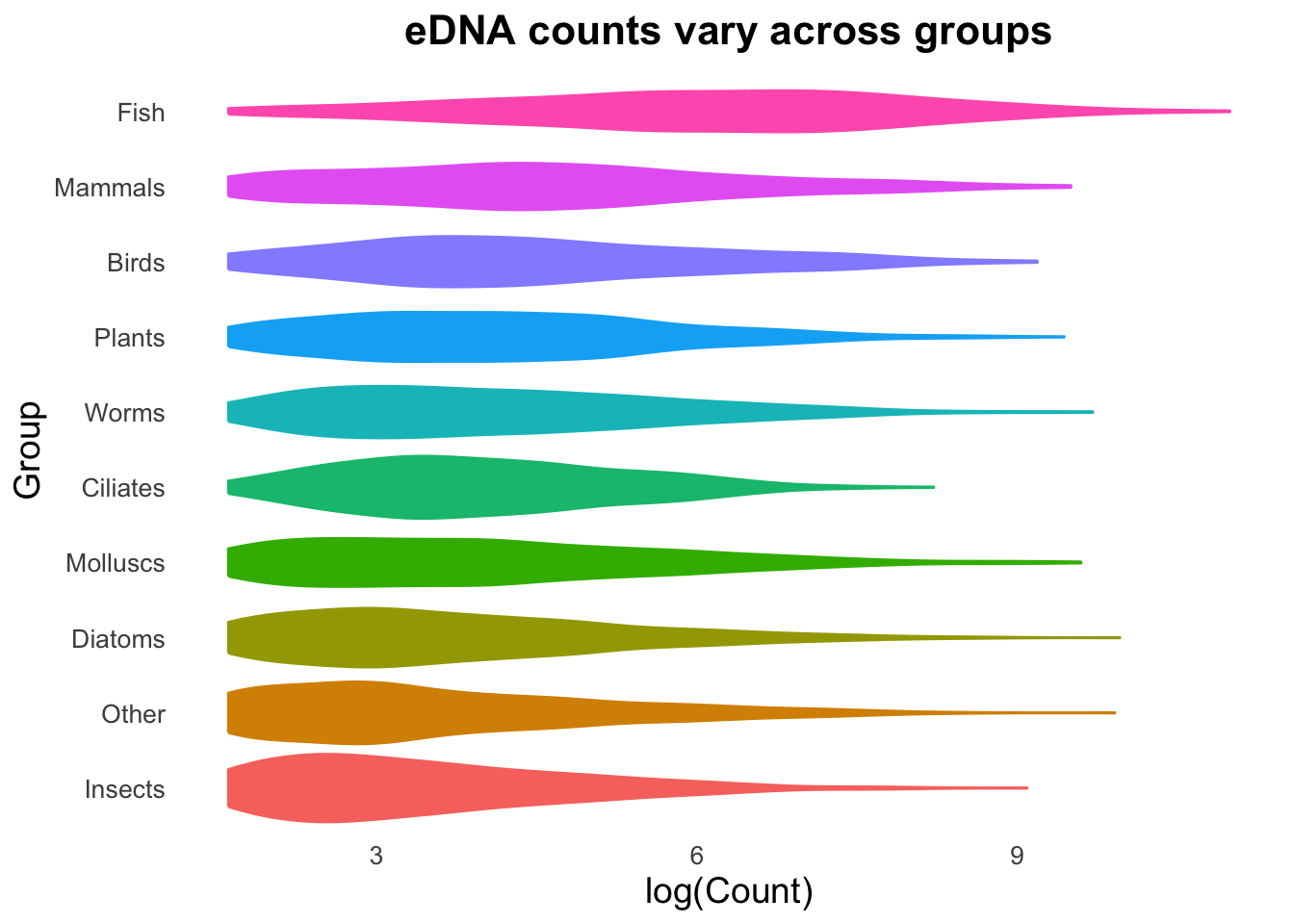
EXERCISE 🧠🏋️♀️ (2 mins)
The function geom_jitter() adds random horizontal movement to our points, which can be controlled with the width argument.
- Try changing the width to:
width = 0.0width = 0.5
width = 1
What do you notice about how the points are spaced?
Do you think one of these widths is better than the others for visualising (and appropriate) for this data? Why or why not?
Want to change the vertical spacing instead? Try the height argument as well as width and see how this changes the plot.
EXERCISE 🧠🏋️♀️ (2 mins)
Now that we have our favourite parameters saved in our groupCounts object, we can test out more geoms for visualising distribution.
Try out these two geoms, what do you think? Do they suit this dataset? Can you fine tune them to make them look better (e.g., with the alpha argument).
geom_violin()geom_density_ridges()
(You may need to run install.packages("ggridges") then library(ggridges))
SOLUTION
groupCounts + geom_violin(aes(colour= Group,fill = Group))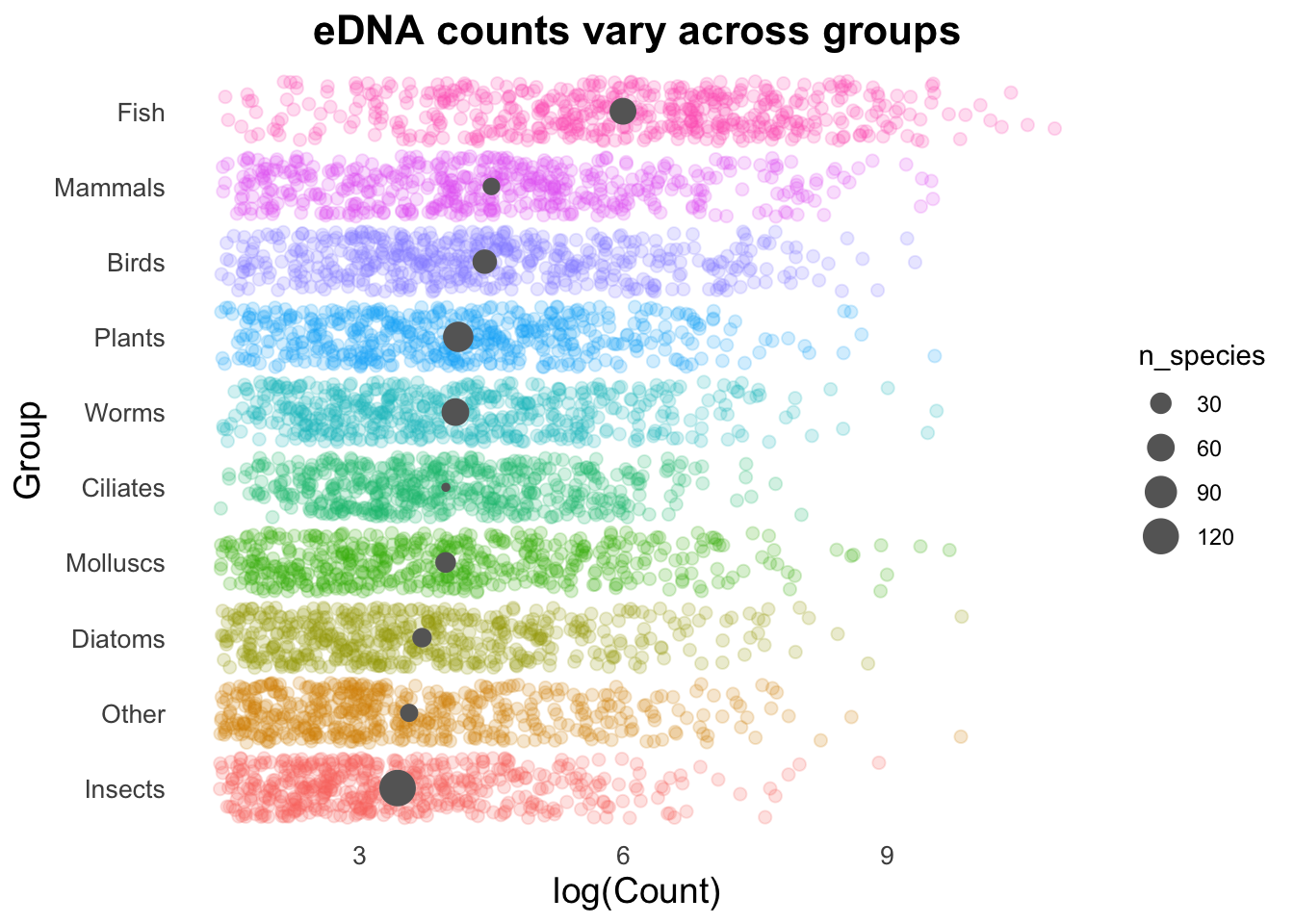
library(ggridges)
groupCounts + geom_density_ridges(aes(colour= Group,fill = Group))Picking joint bandwidth of 0.413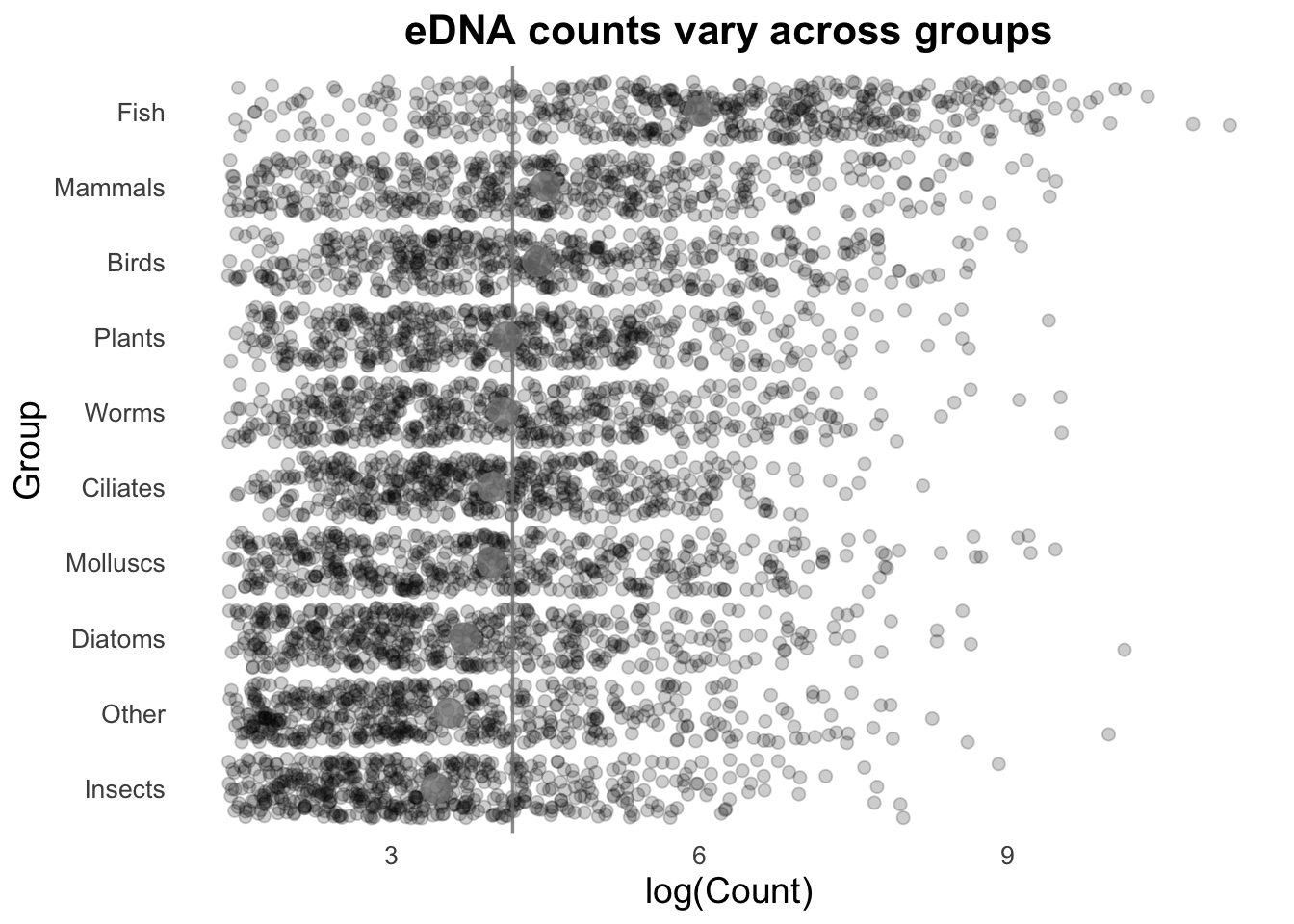
Visualise the data story
There are some really cool things we can do by combining geoms, and here your imagination and creativity is (probably) the limiting factor rather than ggplot. For today, we’ll continue using geom_jitter() for our plot.
Let’s look at how each of our Groups compares to the average (and in this case, we will use mean instead of the median we have been using with our box plots).
First, we will visualise the Group means with the stat_summary() function.
groupCounts +
geom_jitter(size = 2,
alpha = 0.2,
width = 0.2,
aes(colour = Group)) +
stat_summary(fun = mean,
geom = "point",
size = 5,
colour = "gray40")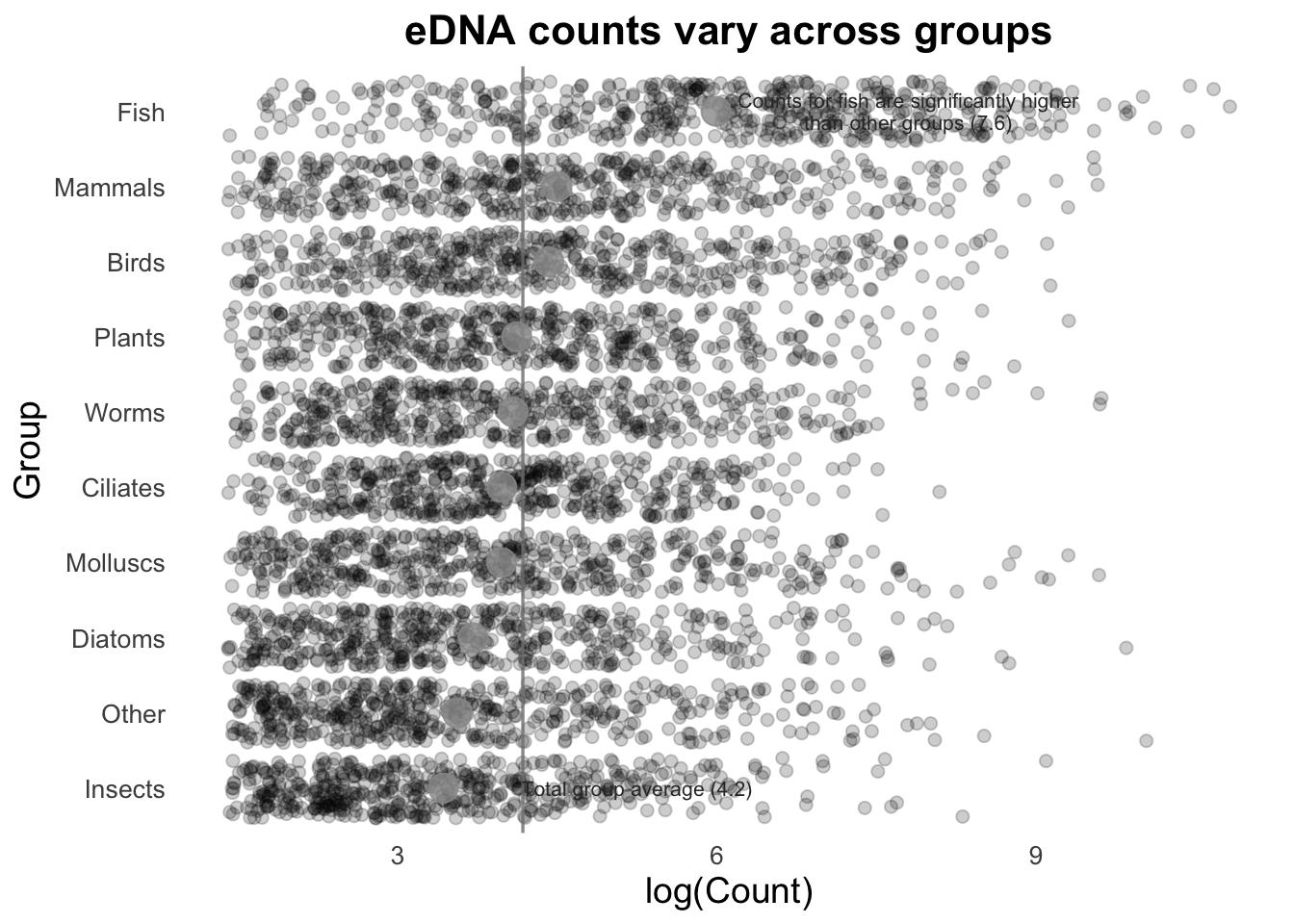
Making use of values
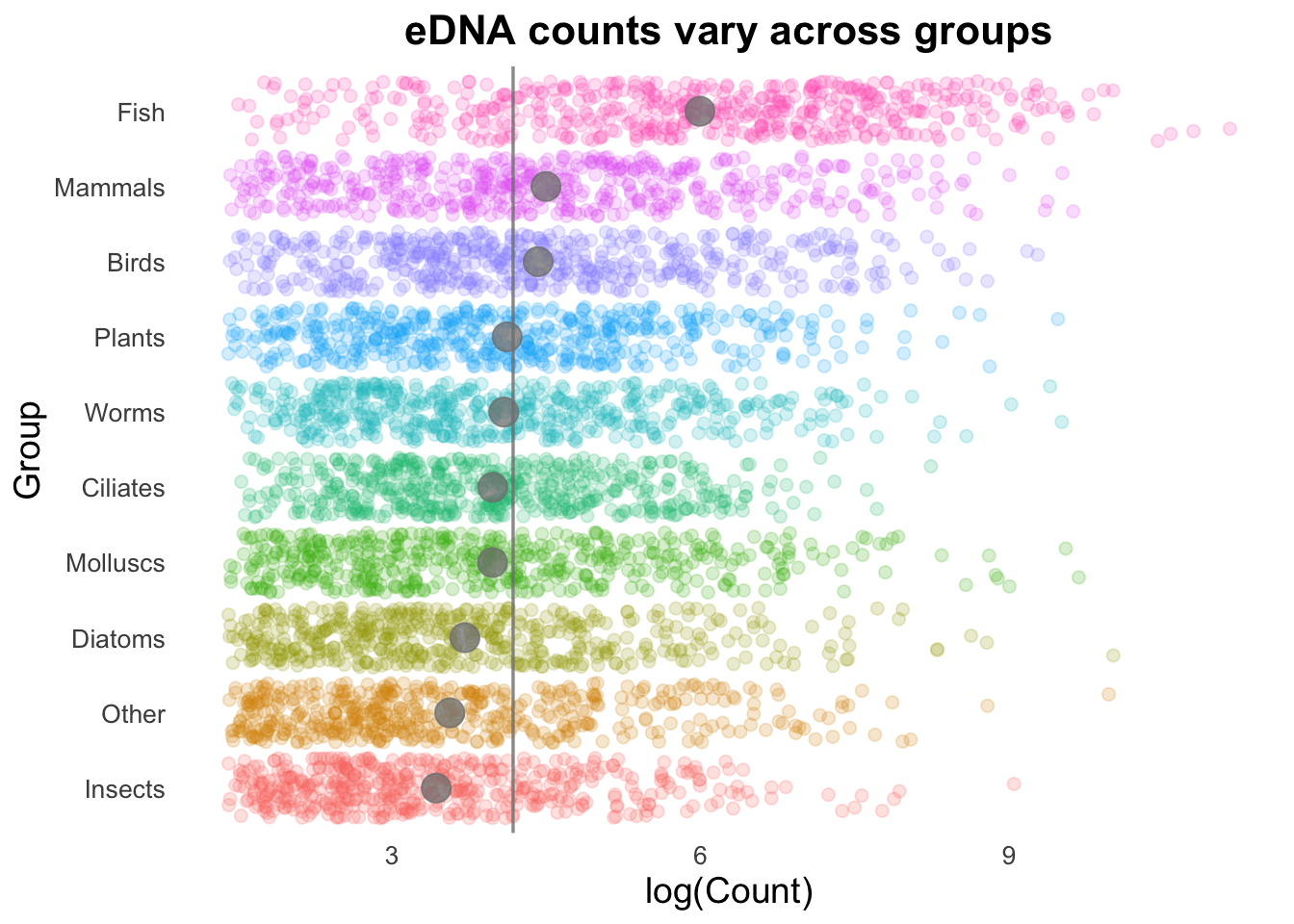
In the code above we have specified the size of the stat_summary() point to be a fixed value (5). In almost all cases within ggplot, you can select dynamic values for arguments e.g., if we had a different number of observations per group, point size could reflect these differences. Making full use of all of these variables leads to very informative plots.
Extra for experts - customise point size
Let’s make the size of the stat_summary points reflect the number of unique species in each group.
Count the number of unique species in each group:
species_counts <- top_group_counts_sorted |>
filter(Rank == "species") |>
group_by(Group) |>
summarise(n_species = n_distinct(Name))Make a new object, joining on the species_counts to our original top_group_counts_sorted object.
plot_data <- top_group_counts_sorted |>
left_join(species_counts, by = "Group")Remake our plot, but now with the size of the stat_summary points reflecting the number of unique species in each group. We also need to add a legend back in on the right, to see what the size of the points means in number of species.
ggplot(plot_data, aes(x = log(Count),
y = Group)) +
labs(title = "eDNA counts vary across groups",
x = "log(Count)",
y = "Group",
size = "Number of unique species") +
theme_minimal() +
theme(
legend.position = "right",
plot.title = element_text(size = 16, face = "bold", hjust = 0.5),
axis.title = element_text(size = 14),
axis.text = element_text(size = 10),
panel.grid = element_blank()
) +
geom_jitter(size = 2,
alpha = 0.2,
width = 0.2,
aes(colour = Group),
show.legend = FALSE) + # alternatively use + guides(colour = "none")
stat_summary(
aes(size = n_species),
fun = mean,
geom = "point",
colour = "gray40"
)
Next, we can add the overall mean for these groups as a vertical line:
# Calculate the average number of Counts across all samples:
top10_group_avg <- mean(log(top_group_counts_sorted$Count))
# Plot:
groupCounts +
geom_vline(aes(xintercept = top10_group_avg),
colour = "gray50",
linewidth = 0.6) +
stat_summary(colour = "gray40",
fun = mean,
geom = "point",
size = 5) +
geom_jitter(size = 2,
alpha = 0.2,
width = 0.2,
aes(colour = Group))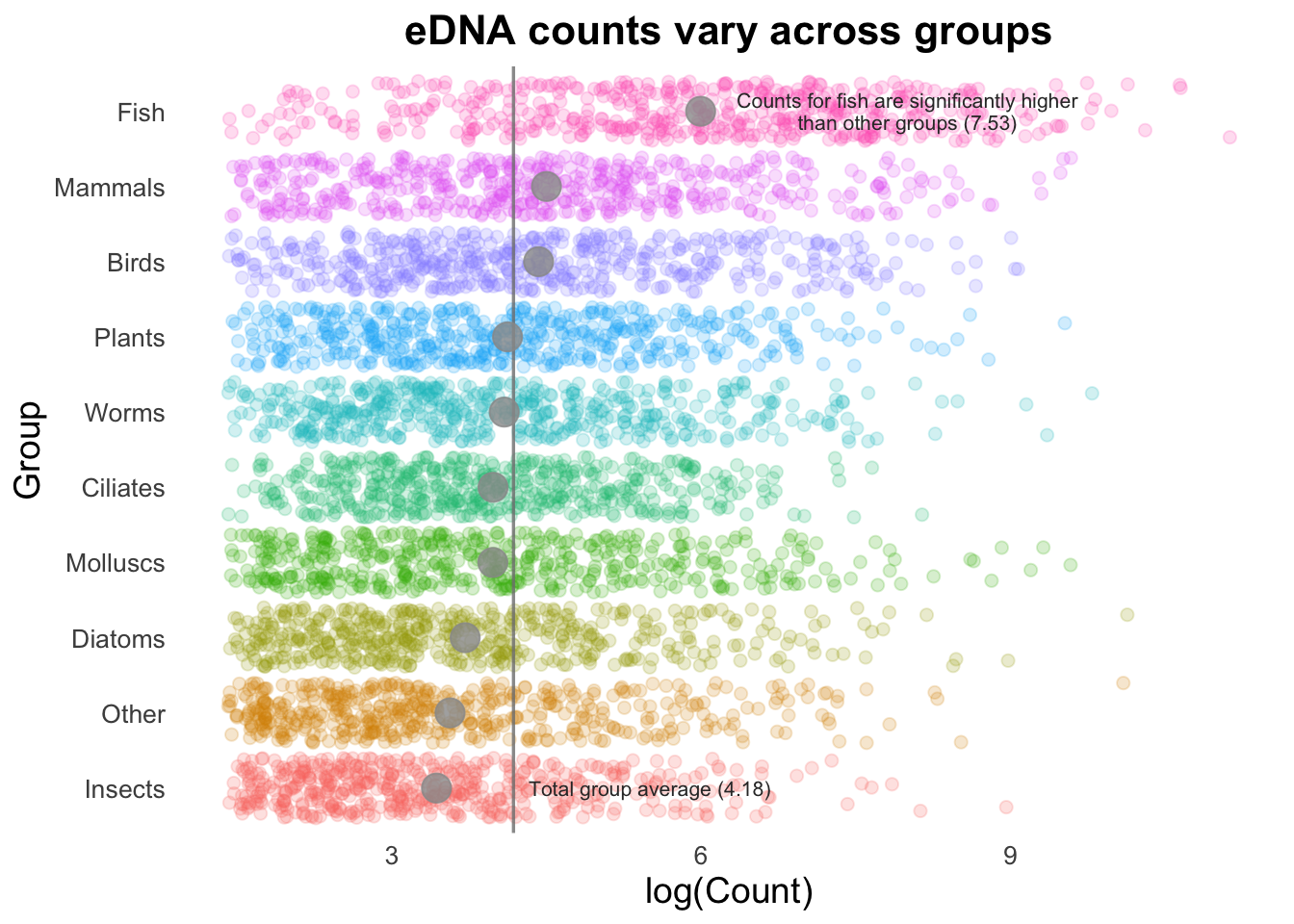
Geom order matters! Note how the summary points (black) are obscured by the coloured points produced by geom_jitter()? We can switch the order so that the jitter points are drawn first, and the summary points placed on top.
groupCounts +
geom_jitter(size = 2,
alpha = 0.2,
width = 0.2,
aes(colour = Group)) +
geom_vline(aes(xintercept = top10_group_avg),
colour = "gray50",
linewidth = 0.6,
alpha = 0.8) +
stat_summary(colour = "gray50",
fun = mean,
geom = "point",
size = 5,
alpha = 0.75)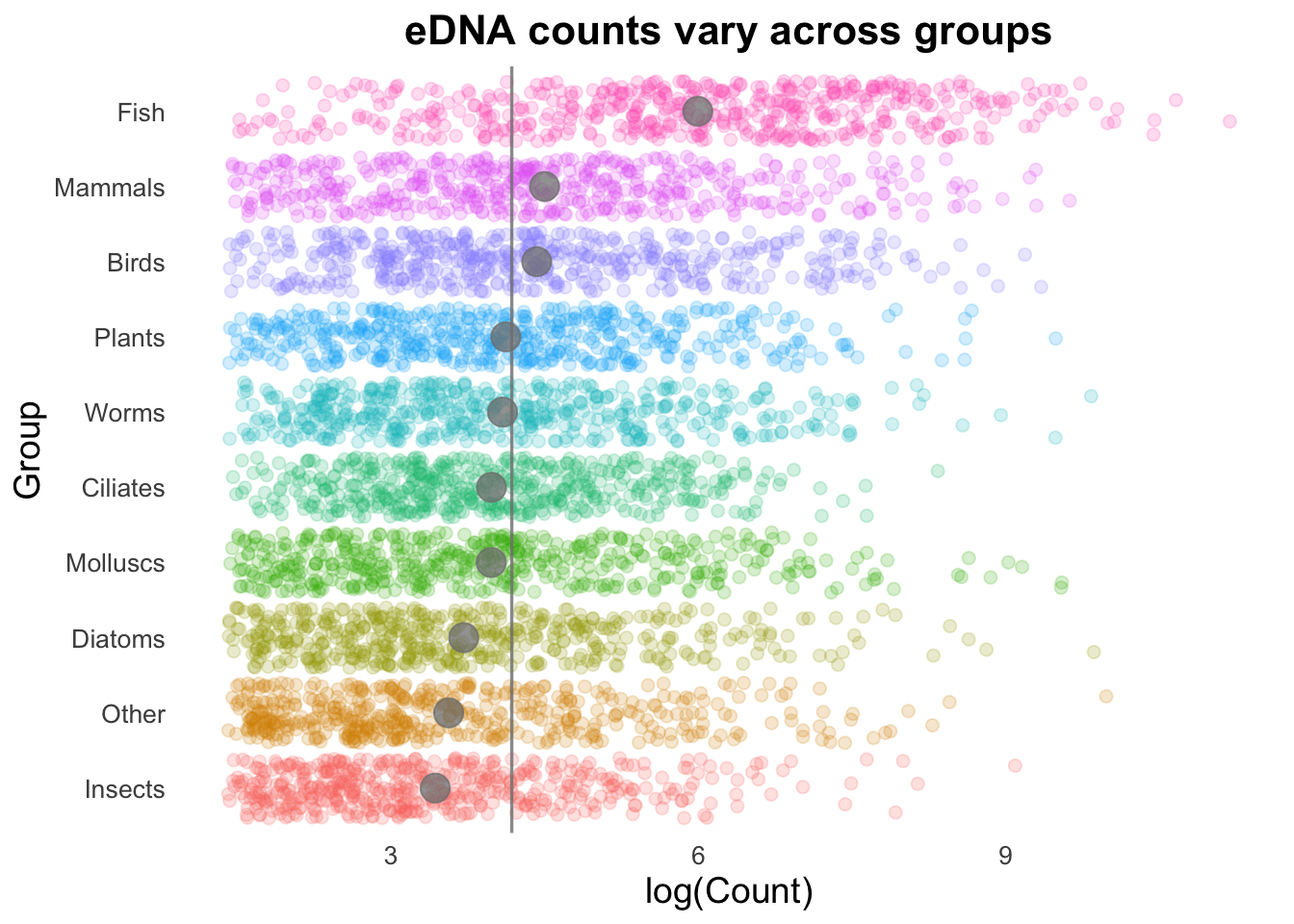
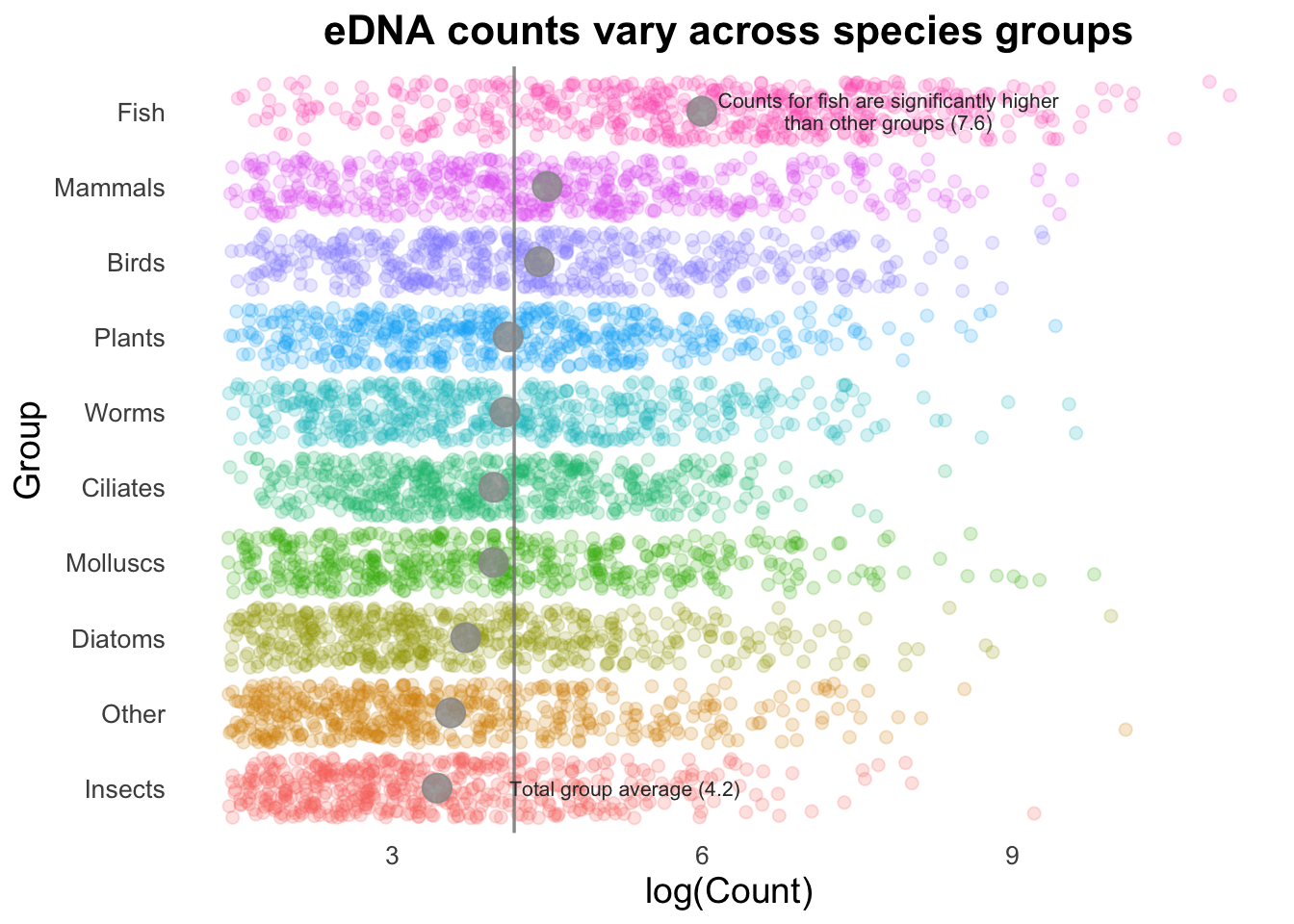
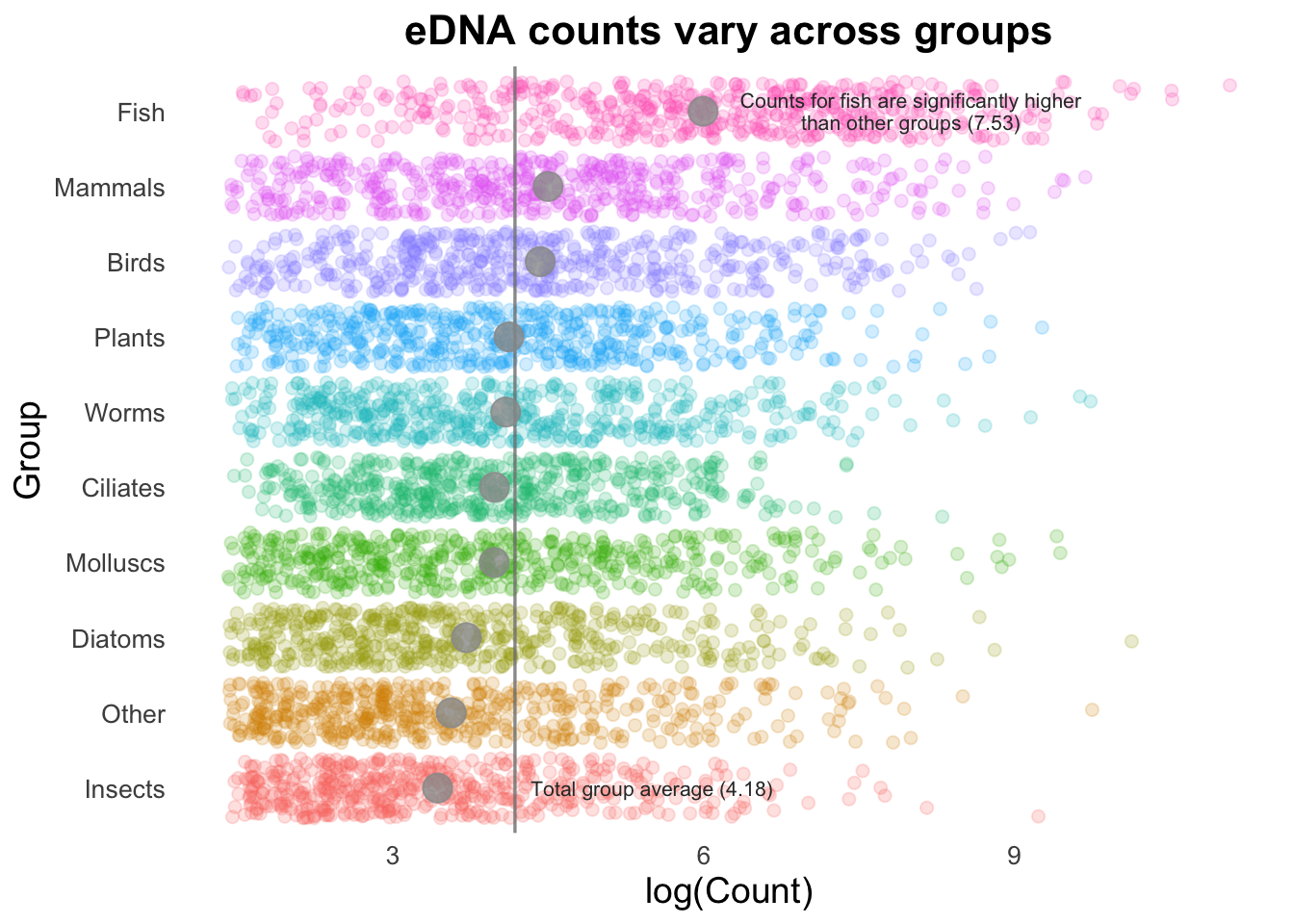
If we are trying to highlight the difference between a Group mean and the global mean, this can be amplified by adding text to our image. (This isn’t necessarily something you would do in many types of plots, but is useful to draw attention to key points e.g., a single gene of interest in a scatter plot).
# Pull out mean of fish group, to use in the text annotation below:
fishMean <- top_group_counts_sorted %>%
filter(Group == "Fish") %>%
summarise(fishMean = log(mean(Count))) %>%
pull(fishMean)
groupCounts +
geom_jitter(size = 2,
alpha = 0.2,
width = 0.2,
aes(colour = Group)) +
geom_vline(aes(xintercept = top10_group_avg),
colour = "gray50",
linewidth = 0.6,
alpha = 0.8) +
stat_summary(colour = "gray60",
fun = mean,
geom = "point",
size = 5,
alpha = 0.8) +
annotate(
"text", x = 8, y = 10, size = 2.8, color = "gray20", lineheight = .9,
label = glue::glue("Counts for fish are significantly higher\nthan other groups ({round(fishMean, 2)})")
) +
annotate(
"text", x = 5.5, y = 1, size = 2.8, color = "gray20",
label = glue::glue("Total group average ({round(top10_group_avg, 2)})")
)
Final CHALLENGE ⚡️
Customise the colours in the above plot to be more aesthetically pleasing.
You can use common palettes such as viridis, pick colours by name, or try choose your own palette from the R graph gallery color palette picker.
Hint: If you use the R graph gallery colour picker option linked above, the library paleteer is not installed on this training environment. But you can chose a palette you like, then hit the download button to get a vector of the hexadecimal codes.
Viridis
We used viridis earlier in the workshop, try adding this line on to the code:
+ scale_color_viridis(discrete = T, option = "mako", direction = -1)Explore the different options using ?scale_colour_viridis.
What other
optionarguments are there?What effect does changing
directionto1or-1have?
Manually add colours
You can also add colours manually for each group using scale_colour_manual(values = c("..."). You’ll need to make sure you have the same number of colours in your vector as number of Groups (n= 10). You can use either the predefined colour names (link earlier ,or type colors() to see all names) or any hexadecimal code in a vector.
+ scale_colour_manual(values = c("coral4","peru", "burlywood","maroon4","violetred4","darkorchid4", "lightslateblue", "midnightblue", "aquamarine4","chartreuse4"))
SOLUTIONS 🌈
There are multiple ways to customise colours in this plot.
You could add:
+ scale_colour_viridis(discrete=T)and you can change the palette options and directions within viridis:
+ scale_color_viridis(discrete = T, direction = -1, option = "magma")or set colours manually:
+ scale_colour_manual(values = c("steelblue", "coral", "darkgreen", "goldenrod", "purple", "cyan", "magenta", "orange", "brown", "pink"))or sample 10 colours at random from the predefined list:
+ scale_colour_manual(values = c(sample(colors(), 10)))Here is a full example:
# save vector of favourite colours as hexadecimal codes
my_colour_palette <- c("#693829FF", "#894B33FF", "#A56A3EFF", "#CFB267FF", "#D9C5B6FF", "#9CA9BAFF", "#5480B5FF", "#3D619DFF", "#405A95FF", "#345084FF")
set.seed(123) #jitter adds randomisation, set seed allows visual reproducibility
groupCounts +
geom_jitter(size = 2,
alpha = 0.4,
width = 0.2,
aes(colour = Group,
)) +
geom_vline(aes(xintercept = top10_group_avg),
colour = "gray50",
linewidth = 0.6) +
stat_summary(colour = "gray20",
fun = mean,
geom = "point",
size = 5,
alpha = 1) +
# change "text" to "label" to put white box around text
annotate(
"label", x = 8, y = 10, size = 2.8, color = "gray20", lineheight = .9,
label = glue::glue("Counts for fish are significantly higher\nthan other groups ({round(fishMean, 2)})")
) +
annotate(
"label", x = 5.5, y = 1, size = 2.8, color = "gray20",
label = glue::glue("Total group average ({round(top10_group_avg, 2)})")
) +
scale_colour_manual(values = my_colour_palette) 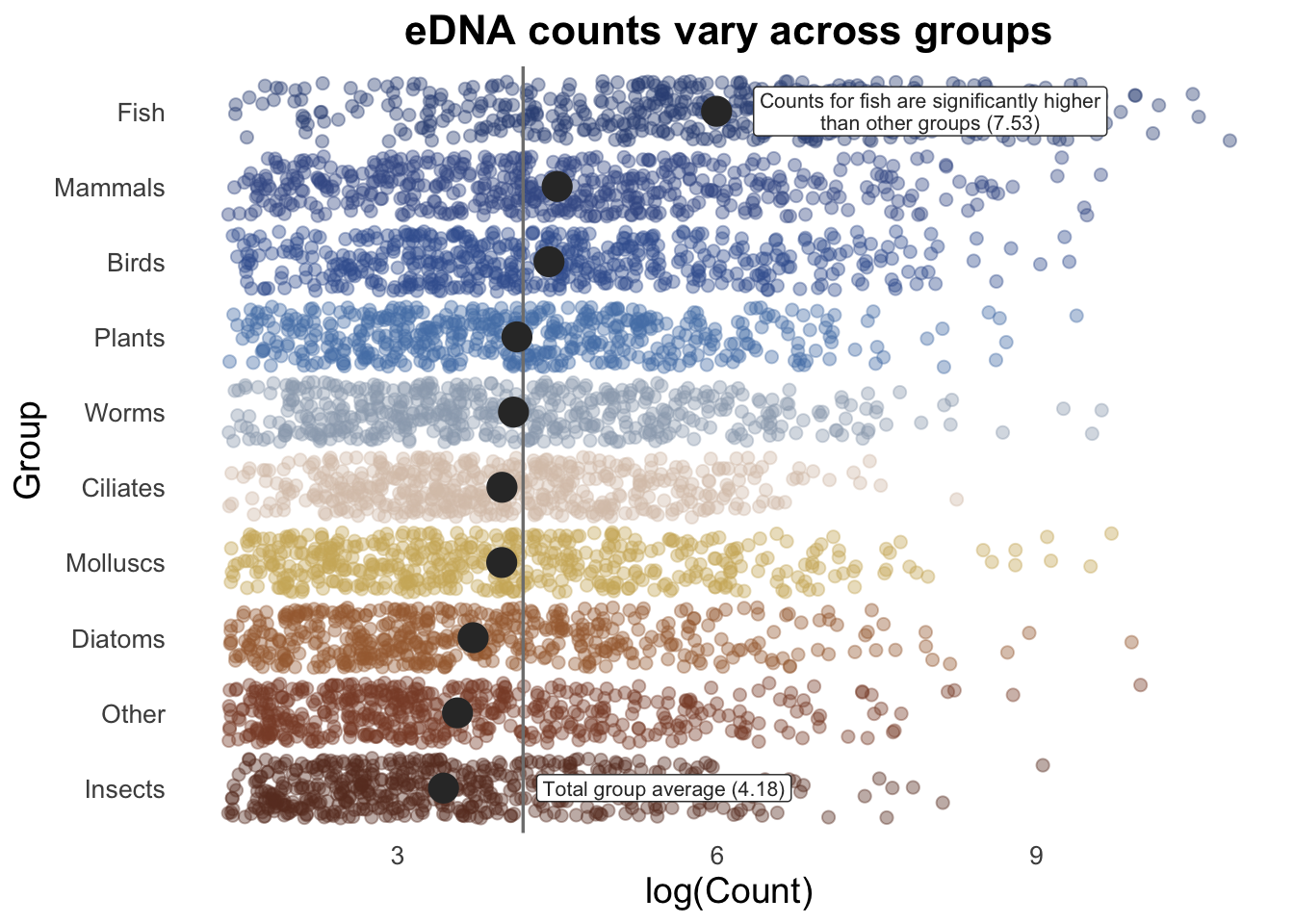
Saving a plot
ggsave() will save the most recently created plot. We can run the code below to save a plot with a given set of dimensions. The default is inches, but we can set this to cm or pixels with the argument units = "cm" or units = "px".
ggsave("edna_plot.png", height = 10, width = 8)Summary
- Save some of the ggplot2 code into an object, to quickly and easily iterate on a plot, letting us sketch out different plot types and styles.
- Use the theme function to save a set of traits that we can use across all of our visualisations, giving us a cohesive look across a single document.
- Geom functions can be combined in different ways to build up a complex chart, while still sticking to our basic template.