BayPass¶
The BayPass manual can be found here.
Citation
Gautier, M. (2015). Genome-wide scan for adaptive divergence and association with population-specific covariates. Genetics, 201(4), 1555-1579. https://doi.org/10.1534/genetics.115.181453
BayPass requires that the allele frequency data be on a population, not an individual basis. The genotyping data file is organized as a matrix with nsnp rows and 2 ∗ npop columns. The row field separator is a space. More precisely, each row corresponds to one marker, and the number of columns is twice the number of populations because each pair of numbers corresponds to each allele (or read counts for PoolSeq experiment) counts in one population.
To generate this population gene count data, we will work with the PLINK file. First, we have to fix the individual ID and population labels, as PLINK has pulled these directly from the VCF, which has no population information. We aim for population in column 1 and individual ID in column 2.
code
cd $DIR/data
awk '{print $2,"\t",$1}' $METADATA > starling_3populations_metadata_POPIND.txt
cd $DIR/analysis/baypass
PLINK=$DIR/data/starling_3populations.plink.ped
#remove first 2 columns
cut -f 3- $PLINK > x.delete
paste $DIR/data/starling_3populations_metadata_POPIND.txt x.delete > starling_3populations.plink.ped
rm x.delete
cp $DIR/data/starling_3populations.plink.map .
cp $DIR/data/starling_3populations.plink.log .
Run the population-based allele frequency calculations.
code
module load PLINK/1.09b6.16
plink --file starling_3populations.plink --allow-extra-chr --freq counts --family --out starling_3populations
Output
PLINK v1.90b6.16 64-bit (19 Feb 2020) www.cog-genomics.org/plink/1.9/
(C) 2005-2020 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to starling_3populations.log.
Options in effect:
--allow-extra-chr
--family
--file starling_3populations.plink
--freq counts
--out starling_3populations
128824 MB RAM detected; reserving 64412 MB for main workspace.
.ped scan complete (for binary autoconversion).
Performing single-pass .bed write (5007 variants, 39 people).
--file: starling_3populations-temporary.bed +
starling_3populations-temporary.bim + starling_3populations-temporary.fam
written.
5007 variants loaded from .bim file.
39 people (0 males, 0 females, 39 ambiguous) loaded from .fam.
Ambiguous sex IDs written to starling_3populations.nosex .
--family: 3 clusters defined.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 39 founders and 0 nonfounders present.
Calculating allele frequencies... done.
Total genotyping rate is 0.723331.
Note: --freq 'counts' modifier has no effect on cluster-stratified report.
--freq: Cluster-stratified allele frequencies (founders only) written to
starling_3populations.frq.strat .
Manipulate file so it has BayPass format, numbers set for PLINK output file, and population number for column count.
code
Now we can run Baypass. It should run for about 5 minutes.
code
Run in R to make the anapod data. First, let us quickly download the utilities we need.
Now let us generate some simulated data based on the parameters calculated from our genetic data.
code
library("ape")
setwd("~/outlier_analysis/analysis/baypass")
source("../../programs/BayPass/baypass_utils.R")
omega <- as.matrix(read.table("starling_3populations_baypass_mat_omega.out"))
pi.beta.coef <- read.table("starling_3populations_baypass_summary_beta_params.out", header = TRUE)
bta14.data <- geno2YN("starling_3populations_baypass.txt")
simu.bta <- simulate.baypass(omega.mat = omega, nsnp = 5000, sample.size = bta14.data$NN, beta.pi = pi.beta.coef$Mean, pi.maf = 0, suffix = "btapods")
q()
We now have the simulated genetic data. We can find the XtX statistic threshold above which we will consider genetic sites an outlier.
code
XtX calibration; get the pod XtX threshold.
code
We compute the 1% threshold for the simulated neutral data.
Your values may be slightly different as the simulated data will not be identical.
Next, we filter the data for the outlier SNPs by identifying those above the threshold.
code
SNP IDs are lost in BayeScan, line numbers are used as signifiers. We have previously created a list of SNPs in VCF and line numbers, which can be found at $DIR/analysis/starling_3populations_SNPs.txt which we will now reuse to generate a list of the outlier SNPS.
code
Now let's find SNPs that are statistically associated with wingspan. To do this, we have to go back to the metadata and compute the average wingspan of each of our populations and place them in a file.
code
setwd("~/outlier_analysis/analysis/baypass")
metadata <- read.table("../../data/starling_3populations_metadata.txt", sep = "\t", header = FALSE)
str(metadata)
pop_metadata <- aggregate(V3 ~ V2, data = metadata, mean)
# Check mean wingspan
pop_metadata[, 2]
write(pop_metadata[, 2], "pop_mean_wingspan.txt")
q()
Now we can run the third and final BayPass job, which will let us know which SNPs are statistically associated with wingspan.
code
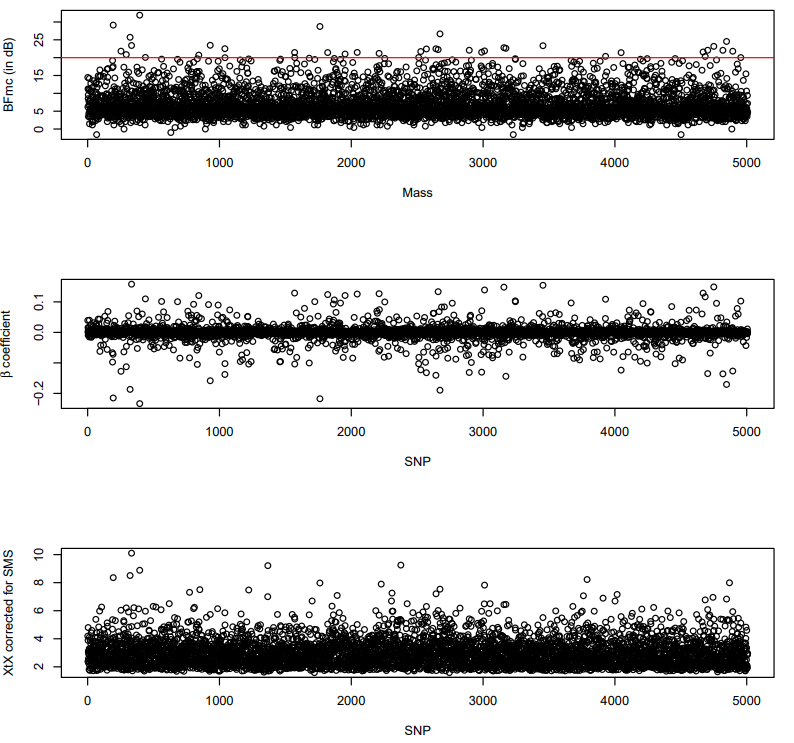
Next we plot the outliers. We are choosing a BF threshold of 20 dB, which indicates "Strong evidence for alternative hypothesis."
code
library(ggplot2)
setwd("~/outlier_analysis/analysis/baypass")
covaux.snp.res.mass <- read.table("starling_3populations_baypass_wing_summary_betai.out", header = T)
covaux.snp.xtx.mass <- read.table("starling_3populations_baypass_summary_pi_xtx.out", header = T)
pdf("Baypass_plots.pdf")
layout(matrix(1:3,3,1))
plot(covaux.snp.res.mass$BF.dB.,xlab="Mass",ylab="BFmc (in dB)")
abline(h=20, col="red")
plot(covaux.snp.res.mass$M_Beta,xlab="SNP",ylab=expression(beta~"coefficient"))
plot(covaux.snp.xtx.mass$M_XtX, xlab="SNP",ylab="XtX corrected for SMS")
dev.off()
Finally, let's generate the list of phenotype-associated SNP IDs.
code
Filter the data sets for SNPS above BFmc threshold. These are out outlier SNPs that are associated with wingspan.