Streams, Redirection and Pipe¶
Lesson Objectives
- To be able to redirect streams of data in Unix.
- Solve problems by piping several Unix commands.
- Command substitution
Bioinformatics data is often text-based and large. This is why Unix’s philosophy of handling text streams is useful in bioinformatics: text streams allow us to do processing on a stream of data rather than holding it all in memory. Handling and redirecting the streams of data is an essential skill in Unix.
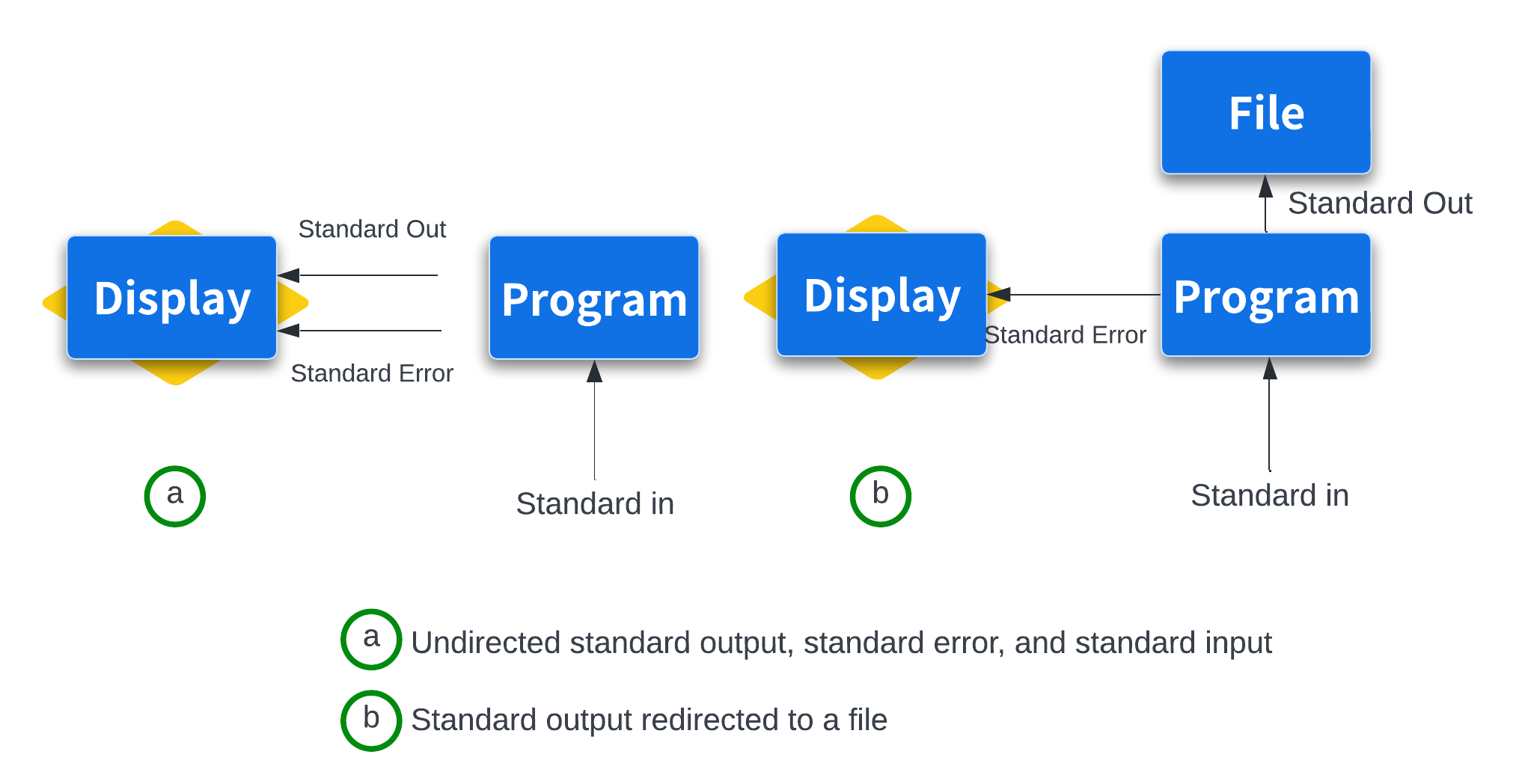
By default, both standard error and standard output of most unix programs go to your terminal screen. We can change this behavior (redirect the streams to a file) by using > or >> operators. The operator > redirects standard output to a file and overwrites any existing contents of the file, whereas >> appends to the file. If there isn’t an existing file, both operators will create it before redirecting output to it.
Output redirection¶
The shell4b_data directory contains the following fasta files:
We can use the cat command to view these files either one at a time:
Recap - cat command to view the content of a file
code
output
>teosinte-branched-1 protein
LGVPSVKHMFPFCDSSSPMDLPLYQQLQLSPSSPKTDQSSSFYCYPCSPP
FAAADASFPLSYQIGSAAAADATPPQAVINSPDLPVQALMDHAPAPATEL
GACASGAEGSGASLDRAAAAARKDRHSKICTAGGMRDRRMRLSLDVARKF
FALQDMLGFDKASKTVQWLLNTSKSAIQEIMADDASSECVEDGSSSLSVD
GKHNPAEQLGGGGDQKPKGNCRGEGKKPAKASKAAATPKPPRKSANNAHQ
VPDKETRAKARERARERTKEKHRMRWVKLASAIDVEAAAASVPSDRPSSN
NLSHHSSLSMNMPCAAA
OR all at once with cat *.fasta
We can also redirect the output to create a new file containing the sequence for both proteins:
Now we have a new file called zea-proteins.fasta. Let's check the contents:
code
output
>teosinte-branched-1 protein
LGVPSVKHMFPFCDSSSPMDLPLYQQLQLSPSSPKTDQSSSFYCYPCSPP
FAAADASFPLSYQIGSAAAADATPPQAVINSPDLPVQALMDHAPAPATEL
GACASGAEGSGASLDRAAAAARKDRHSKICTAGGMRDRRMRLSLDVARKF
FALQDMLGFDKASKTVQWLLNTSKSAIQEIMADDASSECVEDGSSSLSVD
GKHNPAEQLGGGGDQKPKGNCRGEGKKPAKASKAAATPKPPRKSANNAHQ
VPDKETRAKARERARERTKEKHRMRWVKLASAIDVEAAAASVPSDRPSSN
NLSHHSSLSMNMPCAAA
>teosinte-glume-architecture-1 protein
DSDCALSLLSAPANSSGIDVSRMVRPTEHVPMAQQPVVPGLQFGSASWFP
RPQASTGGSFVPSCPAAVEGEQQLNAVLGPNDSEVSMNYGGMFHVGGGSG
GGEGSSDGGT
Capturing error messages
code
>teosinte-branched-1 protein LGVPSVKHMFPFCDSSSPMDLPLYQQLQLSPSSPKTDQSSSFYCYPCSPP FAAADASFPLSYQIGSAAAADATPPQAVINSPDLPVQALMDHAPAPATEL GACASGAEGSGASLDRAAAAARKDRHSKICTAGGMRDRRMRLSLDVARKF FALQDMLGFDKASKTVQWLLNTSKSAIQEIMADDASSECVEDGSSSLSVD GKHNPAEQLGGGGDQKPKGNCRGEGKKPAKASKAAATPKPPRKSANNAHQ VPDKETRAKARERARERTKEKHRMRWVKLASAIDVEAAAASVPSDRPSSN NLSHHSSLSMNMPCAAA cat: mik.fasta: No such file or directory
There are two different types of output there: standard output (the contents of the tb1-protein.fasta file) and standard error (the error message relating to the missing mik.fasta file). If we use the > operator to redirect the output, the standard output is captured, but the standard error is not - it is still printed to the screen. Let's check:
The new file has been created and contains the standard output (contents of the file tb1-protein.fasta):
code
output
>teosinte-branched-1 protein
LGVPSVKHMFPFCDSSSPMDLPLYQQLQLSPSSPKTDQSSSFYCYPCSPP
FAAADASFPLSYQIGSAAAADATPPQAVINSPDLPVQALMDHAPAPATEL
GACASGAEGSGASLDRAAAAARKDRHSKICTAGGMRDRRMRLSLDVARKF
FALQDMLGFDKASKTVQWLLNTSKSAIQEIMADDASSECVEDGSSSLSVD
GKHNPAEQLGGGGDQKPKGNCRGEGKKPAKASKAAATPKPPRKSANNAHQ
VPDKETRAKARERARERTKEKHRMRWVKLASAIDVEAAAASVPSDRPSSN
NLSHHSSLSMNMPCAAA
If we want to capture the standard error we use the (slightly unweildy) 2> operator:
Descriptors
File descriptor 2 represents standard error (other special file descriptors include 0 for standard input and 1 for standard output).
Check the contents:
Reminder :> vs >>
Note that > will overwrite an existing file. We can use >> to add to a file instead of overwriting it:
code
output
>teosinte-branched-1 protein
LGVPSVKHMFPFCDSSSPMDLPLYQQLQLSPSSPKTDQSSSFYCYPCSPP
FAAADASFPLSYQIGSAAAADATPPQAVINSPDLPVQALMDHAPAPATEL
GACASGAEGSGASLDRAAAAARKDRHSKICTAGGMRDRRMRLSLDVARKF
FALQDMLGFDKASKTVQWLLNTSKSAIQEIMADDASSECVEDGSSSLSVD
GKHNPAEQLGGGGDQKPKGNCRGEGKKPAKASKAAATPKPPRKSANNAHQ
VPDKETRAKARERARERTKEKHRMRWVKLASAIDVEAAAASVPSDRPSSN
NLSHHSSLSMNMPCAAA
>teosinte-glume-architecture-1 protein
DSDCALSLLSAPANSSGIDVSRMVRPTEHVPMAQQPVVPGLQFGSASWFP
RPQASTGGSFVPSCPAAVEGEQQLNAVLGPNDSEVSMNYGGMFHVGGGSG
GGEGSSDGGT
The Unix pipe¶
The pipe operator (|) passes the output from one command to another command as input. The following is an example of using a pipe with the grep command.
Steps:
- Remove the header information for the sequence (line starts with ">")
- Highlight any characters in the sequence that are not A, T, C or G.
We will use grep to carry out the first step, and then use the pipe operator to pass the output to a second grep command to carry out the second step.
Here is the full command:
Let's run the code:
code
Output
CCCCAAAGACGGACCAATCCAGCAGCTTCTACTGCTAYCCATGCTCCCCTCCCTTCGCCGCCGCCGACGC
Combining pipes and redirection¶
redirect the standard output of above grep.. command to non-atcg.txt
since we are redirecting to a text file, the --color by itself will not record the colour information. We can achieve this by invoking always flag for --color.i.e..
Using tee to capture intermediate outputs¶
code
The file intermediate-out.txt will contain the output from grep -v "^>" tb1.fasta, but tee also passes that output through the pipe to the next grep command.
Preview - This is to be covered in "Advanced Shell for Bioinformatics"
Pipes and Chains and Long running processes : Exit Status (Programmatically Tell Whether Your Command Worked)
How do you know when they complete? How do you know if they successfully finished without an error? Unix programs exit with an exit status, which indicates whether a program terminated without a problem or with an error. By Unix standards, an exit status of 0 indicates the process ran successfully, and any nonzero status indicates some sort of error has occurred (and hopefully the program prints an understandable error message, too). The exit status isn’t printed to the terminal, but your shell will set its value to a shell variable named $?. We can use the echo command to look at this variable’s value after running a command:
&&), and one operator that runs the next command only if the first completed unsuccessfully (||).
For example, the sequence program1 input.txt > intermediate-results.txt && program2 intermediate-results.txt > results.txt will execute the second command only if previous commands have completed with a successful zero exit status.
By contrast, program1 input.txt > intermediate-results.txt || echo "warning: an error occurred" will print the message if error has occurred.
When a script ends with an exit that has no parameter, the exit status of the script is the exit status of the last command executed in the script (previous to the exit).
Exit Status : using && and ||
To test your understanding of && and ||, we’ll use two Unix commands that do nothing but return either exit success (true) or exit failure (false). Predict and check the outcome of the following commands:
true
echo $?
false
echo $?
true && echo "first command was a success"
true || echo "first command was not a success"
false || echo "first command was not a success"
false && echo "first command was a success"
hint
The $? variable represents the exit status of the previous command.
Answer
Command Substitution¶
Unix users like to have the Unix shell do the work for them. This is why shell expansions like wildcards and brace expansion exist. Another type of useful shell expansion is command substitution. Command substitution runs a Unix command inline and returns the output as a string that can be used in another command. This opens up a lot of useful possibilities. For example, if you want to include the results from executing a command into a text, you can type:
Which is better ?
echo: This is a command that prints text to the standard output.- The text in quotes is what will be printed, with a substitution: "There are ... entries in my FASTA file."
$(...): This is command substitution. It runs the command inside the parentheses and replaces itself with the output of that command.-
grep -c '^@' SRR097977.fastq: This is the command inside the substitution:-c: An option that tells grep to count matching lines instead of printing them'^@': The pattern to search for. In this case, it's looking for lines that start with '@'
-
So, this command will:
- Count how many lines in SRR097977.fastq start with '@'
- Substitute that number into the echo statement
- Print the resulting message!
Another example of using command substitution would be creating dated directories: