RNA-Seq Mapping And Count Data Workflow¶
This material is extracted from the RNA-seq workshop Lesson
Objectives and overall workflow
- To develop a pipeline that does mapping and count the number of reads that mapped overall, then put all these steps into a script.
- Understand and perform the steps involved in RNA-seq mapping and read count.
- Use command line tools to run the pipeline.
RNA-Seq Data Analysis Workflow¶
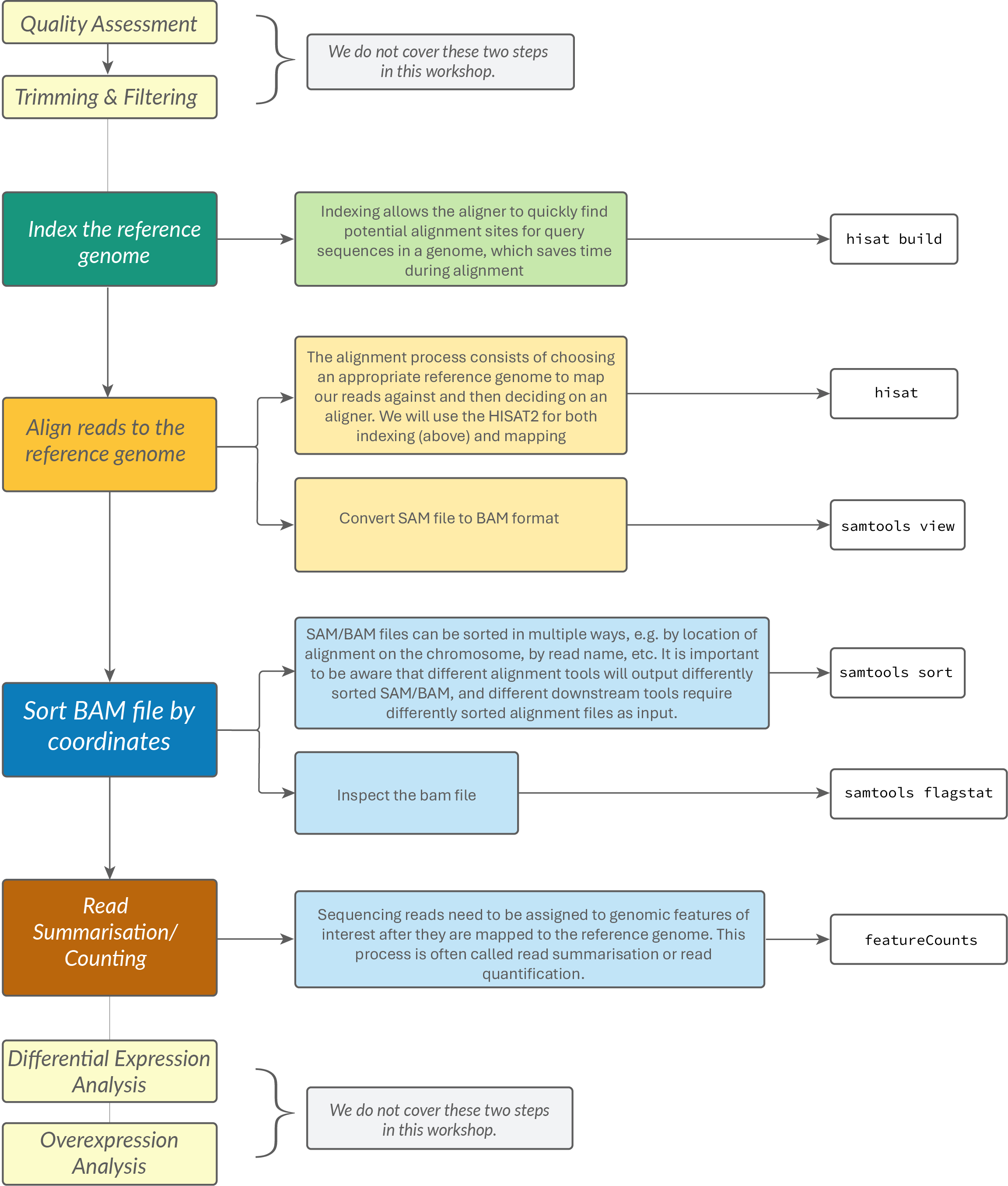
Assumptions¶
- You have already performed trimming and filtering of your reads and saved in a directory called trimmed_reads.
- You have a reference genome saved in a directory called ref_genome.
In this workshop, we have already trimmed the reads and downloaded the reference genome for you. First, it is always good to verify where we are:
Checking to make sure we have the directory and files for the workshop.
Alignment to a reference genome¶
RNA-seq generate gene expression information by quantifying the number of transcripts (per gene) in a sample. This is accomplished by counting the number of transcripts that have been sequenced - the more active a gene is, the more transcripts will be in a sample, and the more reads will be generated from that transcript.
For RNA-seq, we need to align or map each read back to the genome, to see which gene produced it. - Highly expressed genes will generate lots of transcripts, so there will be lots of reads that map back to the position of that transcript in the genome. - The per-gene data we work with in an RNA-seq experiment are counts: the number of reads from each sample that originated from that gene.
Preparation of the genome¶
To be able to map (align) sequencing reads on the genome, the genome needs to be indexed first. In this workshop we will use HISAT2.
script
Output -Saccharomyces_cerevisiae.R64-1-1.99.gtf Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa
# index file:
hisat2-build -p 4 -f Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa Saccharomyces_cerevisiae.R64-1-1.dna.toplevel
Output
Saccharomyces_cerevisiae.R64-1-1.99.gtf Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.4.ht2 Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.8.ht2
Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.1.ht2 Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.5.ht2 Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa
Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.2.ht2 Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.6.ht2
Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.3.ht2 Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.7.ht2
Arguments
- -p number of threads
- -f fasta file
How many files were created during the indexing process?
Alignment on the genome¶
Now that the genome is prepared, sequencing reads can be aligned.
Information required
- Where the sequence information is stored (e.g. fastq files ...) ?
- What kind of sequencing: Single End or Paired end ?
- Where are the indexes and the genome stored?
- Where will the mapping files be stored?
- Now, lets move one folder up (into the rna_seq folder):
Let's map one of our sample to the reference genome:
Let's use a for loop to process our samples:
script
Output -SRR014335-chr1.fastq SRR014336-chr1.fastq SRR014337-chr1.fastq SRR014339-chr1.fastq SRR014340-chr1.fastq SRR014341-chr1.fastq
for filename in *
do
base=$(basename ${filename} .fastq)
hisat2 -p 4 -x ../ref_genome/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel -U $filename -S ../Mapping/${base}.sam --summary-file ../Mapping/${base}_summary.txt
done
Output
125090 reads; of these:
125090 (100.00%) were unpaired; of these:
15108 (12.08%) aligned 0 times
88988 (71.14%) aligned exactly 1 time
20994 (16.78%) aligned >1 times
87.92% overall alignment rate
125936 reads; of these:
125936 (100.00%) were unpaired; of these:
14300 (11.35%) aligned 0 times
90061 (71.51%) aligned exactly 1 time
21575 (17.13%) aligned >1 times
88.65% overall alignment rate
143638 reads; of these:
143638 (100.00%) were unpaired; of these:
28334 (19.73%) aligned 0 times
94006 (65.45%) aligned exactly 1 time
21298 (14.83%) aligned >1 times
80.27% overall alignment rate
80820 reads; of these:
80820 (100.00%) were unpaired; of these:
8399 (10.39%) aligned 0 times
60306 (74.62%) aligned exactly 1 time
12115 (14.99%) aligned >1 times
89.61% overall alignment rate
80453 reads; of these:
80453 (100.00%) were unpaired; of these:
8545 (10.62%) aligned 0 times
60560 (75.27%) aligned exactly 1 time
11348 (14.11%) aligned >1 times
89.38% overall alignment rate
78306 reads; of these:
78306 (100.00%) were unpaired; of these:
8201 (10.47%) aligned 0 times
59517 (76.01%) aligned exactly 1 time
10588 (13.52%) aligned >1 times
89.53% overall alignment rate
Arguments
- -x The basename of the index for the reference genome.
- -U Comma-separated list of files containing unpaired reads to be aligned
- -S File to write SAM alignments to. By default, alignments are written to the “standard out” or “stdout” filehandle
Now we can explore our SAM files.
script
Converting SAM files to BAM files¶
The SAM file is a tab-delimited text file that contains information for each individual read and its alignment to the genome. While we do not have time to go into detail about the features of the SAM format, the paper by Heng Li et al. provides a lot more detail on the specification.
The compressed binary version of SAM is called a BAM file. We use this version to reduce size and to allow for indexing, which enables efficient random access of the data contained within the file.
A quick look into the sam file¶
Hint
Hit q to exit the less interactive display.
.sam header & format
The file begins with a header, which is optional. The header is used to describe the source of data, reference sequence, method of alignment, etc., this will change depending on the aligner being used. Following the header is the alignment section. Each line that follows corresponds to alignment information for a single read. Each alignment line has 11 mandatory fields for essential mapping information and a variable number of other fields for aligner specific information. An example entry from a SAM file is displayed below with the different fields highlighted.
We will convert the SAM file to BAM format using the samtools program with the view command and tell this command that the input is in SAM format (-S) and to output BAM format (-b):
script
Next we sort the BAM file using the sort command from samtools. -o tells the command where to write the output.
Note
SAM/BAM files can be sorted in multiple ways, e.g. by location of alignment on the chromosome, by read name, etc. It is important to be aware that different alignment tools will output differently sorted SAM/BAM, and different downstream tools require differently sorted alignment files as input.
script
We can use samtools to learn more about the bam file as well.
Some stats on your mapping:¶
script
Output
156984 + 0 in total (QC-passed reads + QC-failed reads)
31894 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
136447 + 0 mapped (86.92% : N/A)
0 + 0 paired in sequencing
0 + 0 read1
0 + 0 read2
0 + 0 properly paired (N/A : N/A)
0 + 0 with itself and mate mapped
0 + 0 singletons (N/A : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
Read Summarization¶
Sequencing reads often need to be assigned to genomic features of interest after they are mapped to the reference genome. This process is often called read summarization or read quantification. Read summarization is required by a number of downstream analyses such as gene expression analysis and histone modification analysis. The output of read summarization is a count table, in which the number of reads assigned to each feature in each library is recorded.
Counting¶
- We need to do some counting!
- Want to generate count data for each gene (actually each exon) - how many reads mapped to each exon in the genome, from each of our samples?
- Once we have that information, we can start thinking about how to determine which genes were differentially expressed in our study.
Subread and FeatureCounts¶
- The featureCounts tool from the Subread package can be used to count how many reads aligned to each genome feature (exon).
- We need to specify the annotation information (.gtf file).
You can process all the samples at once:
script
Output/home/$USER/scripting_workshop/rna_seq
featureCounts -a ../ref_genome/Saccharomyces_cerevisiae.R64-1-1.99.gtf -o ./yeast_counts.txt -T 2 -t exon -g gene_id ../Mapping/*sorted.bam
Hint
If you encounter an error along the lines of ERROR: invalid parameter:.., first check that your current working directory is Counts. (it's all about location, location, location 😉 )
Output
========== _____ _ _ ____ _____ ______ _____
===== / ____| | | | _ \| __ \| ____| /\ | __ \
===== | (___ | | | | |_) | |__) | |__ / \ | | | |
==== \___ \| | | | _ <| _ /| __| / /\ \ | | | |
==== ____) | |__| | |_) | | \ \| |____ / ____ \| |__| |
========== |_____/ \____/|____/|_| \_\______/_/ \_\_____/
v2.0.7
//========================== featureCounts setting ===========================\\
|| ||
|| Input files : 6 BAM files ||
|| ||
|| SRR014335-chr1_sorted.bam ||
|| SRR014336-chr1_sorted.bam ||
|| SRR014337-chr1_sorted.bam ||
|| SRR014339-chr1_sorted.bam ||
|| SRR014340-chr1_sorted.bam ||
|| SRR014341-chr1_sorted.bam ||
|| ||
|| Output file : yeast_counts.txt ||
|| Summary : yeast_counts.txt.summary ||
|| Paired-end : no ||
|| Count read pairs : no ||
|| Annotation : Saccharomyces_cerevisiae.R64-1-1.99.gtf (GTF) ||
|| Dir for temp files : . ||
|| ||
|| Threads : 2 ||
|| Level : meta-feature level ||
|| Multimapping reads : not counted ||
|| Multi-overlapping reads : not counted ||
|| Min overlapping bases : 1 ||
|| ||
\\============================================================================//
//================================= Running ==================================\\
|| ||
|| Load annotation file Saccharomyces_cerevisiae.R64-1-1.99.gtf ... ||
|| Features : 7507 ||
|| Meta-features : 7127 ||
|| Chromosomes/contigs : 17 ||
|| ||
|| Process BAM file SRR014335-chr1_sorted.bam... ||
|| Single-end reads are included. ||
|| Total alignments : 161172 ||
|| Successfully assigned alignments : 58373 (36.2%) ||
|| Running time : 0.00 minutes ||
|| ||
|| Process BAM file SRR014336-chr1_sorted.bam... ||
|| Single-end reads are included. ||
|| Total alignments : 163348 ||
|| Successfully assigned alignments : 59351 (36.3%) ||
|| Running time : 0.00 minutes ||
|| ||
|| Process BAM file SRR014337-chr1_sorted.bam... ||
|| Single-end reads are included. ||
|| Total alignments : 179255 ||
|| Successfully assigned alignments : 61791 (34.5%) ||
|| Running time : 0.00 minutes ||
|| ||
|| Process BAM file SRR014339-chr1_sorted.bam... ||
|| Single-end reads are included. ||
|| Total alignments : 107272 ||
|| Successfully assigned alignments : 47147 (44.0%) ||
|| Running time : 0.00 minutes ||
|| ||
|| Process BAM file SRR014340-chr1_sorted.bam... ||
|| Single-end reads are included. ||
|| Total alignments : 104274 ||
|| Successfully assigned alignments : 47473 (45.5%) ||
|| Running time : 0.00 minutes ||
|| ||
|| Process BAM file SRR014341-chr1_sorted.bam... ||
|| Single-end reads are included. ||
|| Total alignments : 99253 ||
|| Successfully assigned alignments : 46614 (47.0%) ||
|| Running time : 0.00 minutes ||
|| ||
|| Write the final count table. ||
|| Write the read assignment summary. ||
|| ||
|| Summary of counting results can be found in file "./yeast_counts.txt.summ ||
|| ary" ||
|| ||
\\============================================================================//
yeast_counts.txt yeast_counts.txt.summary
Arguments:
- -a Name of an annotation file. GTF/GFF format by default.
- -o Name of output file including read counts
- -T Specify the number of threads/CPUs used for mapping. 1 by default.
- -t Specify feature type in GTF annotation. 'exon' by default. Features used for read counting will be extracted from annotation using the provided value.
- -g Specify attribute type in GTF annotation. 'gene_id' by default. Meta-features used for read counting will be extracted from annotation using the provided value.
Group Exercise 3.1
Now, let's work together in our groups to create an RNA-seq mapping and count script.
- You will be assigned to breakout rooms
- One person from the room must volunteer to share their screen as the group works together to compile the script
- It is safe to compile and submit the script from
/rna_seqparent directory as it will override the existing results from the above steps. Applications used in this pipeline will obey the "override" by default. However, some applications will demand the existing outputs to be deleted or use a provided flag such as--override(if it is available as a function of the application) - On the other hand, how about we bring more structure to "results/outputs" ? .i.e. Perhaps create a results directory for
sam,bamandcountssub-directories than creating/Mapping(for both sam and bam) and/Counts? (Similar to what we have done in Variant calling pipeline) - Also, the above testing was done with three separate
forloops. Is it possible to bring them altogether under oneforloop ?
🎊
At this stage we have mastered the art of writing scripts, instead of running them on the command line, let us now run them on HPC.