Automating a Variant Calling Workflow¶
Aim
- Put all the steps from the previous lesson into a script.
Variant calling workflow¶
Remember our variant calling workflow has the following steps:
- Index the reference genome for use by bwa and samtools.
- Align reads to reference genome.
- Convert the format of the alignment to sorted BAM, with some intermediate steps.
- Calculate the read coverage of positions in the genome.
- Detect the single nucleotide variants (SNVs).
- Filter and report the SNVs in VCF (variant calling format).
Let's start with creating a new directory as our script working space and copy all the required resources.
Output -/home/$USER/scripting_workshop
#Don't forget the . at the end of the line
cp -r ../variant_calling/ref_genome ../variant_calling/trimmed_reads .
Output - ref_genome trimmed_reads
Now we are ready to start building the script.
Prefer Jupyter file over nano ?
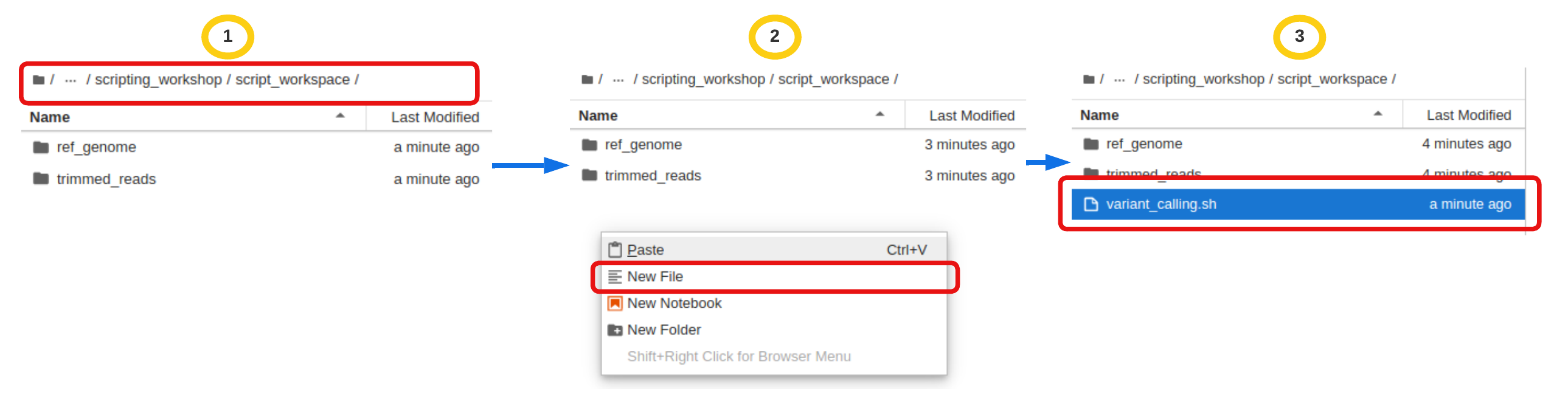
You are welcome to choose Jupyter interactive file option over nano. If this is your preferred option, below are the instructions on how to create a file
- Make sure to navigate the left file explorer to correct path
- Right click on the explorer to open the menu and choose
New file - Rename the file as
variant_calling.sh
In the text editor, type the commands
Shell variables
A variable is a character string to which we assign a value. The value assigned could be a number, text, filename, device, or any other type of data. A variable is nothing more than a pointer to the actual data. The shell enables you to create, assign, and delete variables.
Adding executable permissions
The way the script is written means we have to indicate which program to use whenever we are running it. To be able to run without calling bash, we need to change the script permissions.
script
Output -rw-rw----+ 1 userID nesi02659 1411 Apr 16 13:53 variant_calling.sh
Output - -rwxrw----+ 1 userID nesi02659 1411 Apr 16 13:53 variant_calling.sh
- note colour change on the script filename
Now we can execute the script without calling bash
In the Next Lesson, we will do a similar pipeline, but with RNAseq data.