nf-core/sarek¶
Objectives
- Understand the Sarek pipeline structure and default usage
- Understand the levels of customisation available for nf-core pipelines
- Use the nf-core documentation to select appropriate parameters for a run command
- Write and run a nf-core sarek command on the command line
- Explore pipeline deployment and outputs
The Sarek pipeline¶
nf-core/sarek is a pipeline designed to detect variants on whole genome or targeted sequencing data. Initially designed for Human, and Mouse, it can work on any species with a reference genome. Sarek can also handle tumour/normal pairs and could include additional relapses.
The pipeline makes use of Docker/Singularity containers, making installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies.
Depending on the options and samples provided, the pipeline can currently perform the following:
- Form consensus reads from UMI sequences (
fgbio) - Sequencing quality control and trimming (
FastQC,fastp) - Map Reads to Reference (
BWA-memorBWA-mem2ordragmap) - Process BAM file (
GATK MarkDuplicates,GATK BaseRecalibrator,GATK ApplyBQSR) - Summarise alignment statistics (
samtools stats,mosdepth) - Variant calling (enabled by
--tools, see compatibility):HaplotypeCaller,freebayes,mpileup,Strelka2,DeepVariant,Mutect2,Manta,TIDDIT,ASCAT,Control-FREEC,CNVkit, and / orMSIsensor-pro
- Variant filtering and annotation (
SnpEff,Ensembl VEP) - Summarise and represent QC (
MultiQC)
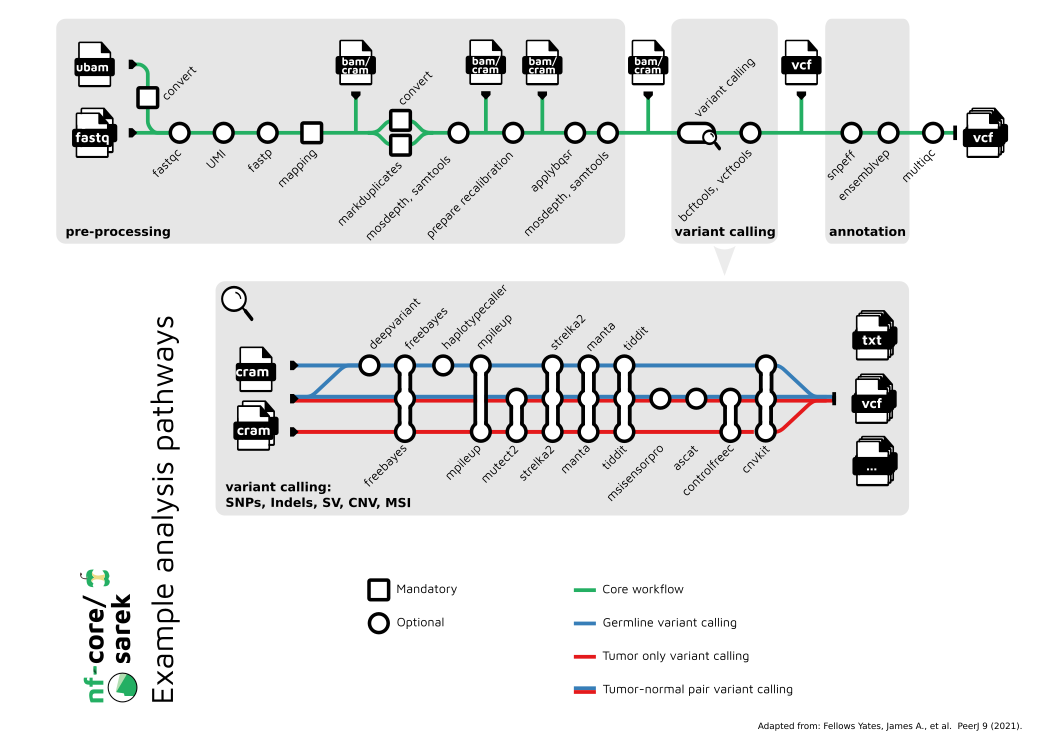
nf-core pipelines are frequently represented as subway maps. The nf-core/sarek subway map is shown below and is a good place to start when first understanding how the pipeline works.
Download the Sarek pipeline¶
There are multiple ways you can download and store a copy of a nf-core pipeline.
Firstly, you could use the nextflow pull command. By default, if you you the nextflow run command to execute a pipeline from github it will also pull the pipeline. In both of these cases the pipeline will be stored in a hidden directory in your home directory.
Secondly, you could clone a copy of the pipeline using the standard git clone command, e.g., git clone https://github.com/nf-core/sarek.git. This will download the pipeline to your current working directory.
Finally, you could use the nf-core download utility to download a copy of the pipeline. This will give the the option to download the pipeline code, the required singularity images, and the institutional configs from the nf-core github repository. This method can be especially helpful if you are working offline and want to move all of the pipeline code and tooling to a different machine.
Getting started¶
All nf-core pipelines are provided with comprehensive documentation that explain what the default pipeline structure entails and options for customising this based on your needs. It is important to remember that nf-core pipelines typically do not include all possible tool parameters. Instead, they provide a sensible set of parameters that are suitable for most use cases.
The number and type of parameters an nf-core pipeline accepts differ between pipelines. The recommended (typical) run command and all the parameters available for the nf-core/sarek pipeline can be viewed using the --help flag:
Revision 3.2.3
The Sarek pipeline is always improving but we want to ensure that the results of this workshop are reproducible. To ensure this, we will use a specific version (3.2.3) of the pipeline and the revision flag (-r).
At the top of the help output, you will see the recommended run command:
It outlines the requirement for three things:
- An input samplesheet (
--input) - A reference genome (
--genome) - A software management profile (
--profile)
Hyphens matter
Nextflow-specific parameters use one (-) hyphen, whereas pipeline-specific parameters use two (--).
More information about Sarek¶
There is extensive information about nf-core pipelines on the nf-core website. The dedicated Sarek pipeline page is the best resource for information about the pipeline and how to execute it.
If you have specific questions that are not included in the documentation you can join the nf-core Slack workspace and ask in the #sarek channel.
Testing a pipeline¶
Before running a pipeline on your own data, it is a good idea to test the pipeline on a small dataset. This allows you to check that the pipeline is working as expected without having to wait for a long time for the pipeline to complete.
The test profile will run the pipeline on a small test dataset that is included with the pipeline code.
Exercise
Check that Sarek is working by running the pipeline with the test profile.
Test data
The --input and --genome parameters are not required when using the test profile. The test data and small reference files are included with the pipeline code and are automatically used when the test profile is specified.
Key points
- nf-core pipelines are provided with sensible default settings and required inputs.
- The
--helpflag can be used to view the recommended run command and all available parameters. - The
testprofile can be used to show that a pipeline is working as expected.