Configure nf-core pipelines¶
Objectives
- Describe the features of an nf-core pipeline.
- Use command line options to alter a workflow's execution.
- Customize an nf-core pipeline using a configuration file.
- Employ a profile to switch configurations.
Pipeline structure¶
nf-core pipelines follow a set of best practices and standardised conventions. nf-core pipelines start from a common template and follow the same structure. Although you won't need to edit code in the pipeline project directory, having a basic understanding of the project structure and some core terminology will help you understand how to configure its execution.
Nextflow DSL2 workflows are built up of subworkflows and modules that are stored as separate .nf files.
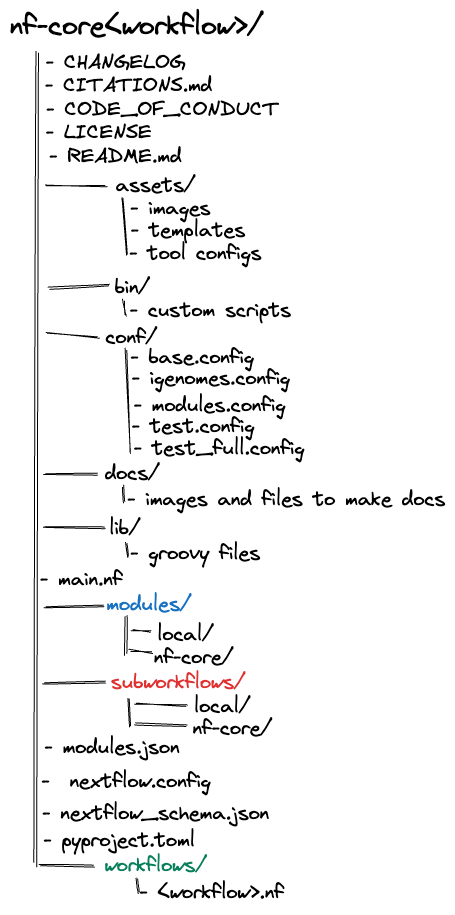
Most nf-core pipelines consist of a combination of subworkflows and modules.
A single workflow file (there are a few exceptions). This is the main <workflow>.nf file that is used to bring everything else together. Instead of having one large monolithic script, it is broken up into a combination of subworkflows and modules.
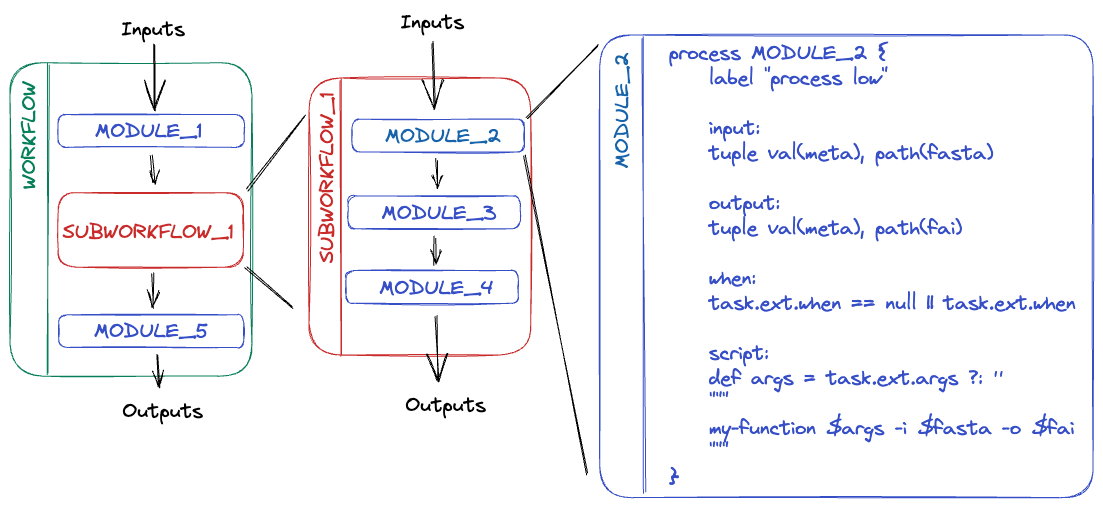
A subworkflow is a group of modules that are used in combination with each other and have a common purpose. For example, the SAMTOOLS_STATS, SAMTOOLS_IDXSTATS, and SAMTOOLS_FLAGSTAT modules are all included in the BAM_STATS_SAMTOOLS subworkflow.
Subworkflows improve pipeline readability and help with the reuse of modules within a pipeline. Within a nf-core pipeline, a subworkflow can be an nf-core subworkflow or a local subworkflow. Like an nf-core pipeline, an nf-core subworkflow is developed by the community and is shared in the nf-core subworkflows GitHub repository. Local subworkflows are pipeline specific and are not shared in the nf-core subworkflows repository.
A module is a wrapper for a process, the basic processing primitive to execute a user script. It can specify directives, inputs, outputs, when statements, and a script block.
Most modules will execute a single tool in the script block and will make use of the directives, inputs, outputs, and when statements dynamically. Like subworkflows, modules can also be developed and shared in the nf-core modules GitHub repository or stored as a local module. All modules from the nf-core repository are version controlled and tested to ensure reproducibility. Local modules are pipeline specific and are not shared in the nf-core modules repository.
Configuration¶
Each nf-core pipeline comes with a set of "sensible defaults". While the defaults are a great place to start, you will almost certainly want to modify these to fit your own purposes and system requirements.
You do not need to edit the pipeline code to configure nf-core pipelines.
When a pipeline is launched, Nextflow will look for configuration files in several locations. As each source can contain conflicting settings, the sources are ranked to decide which settings to apply. Configuration sources are reported below and listed in order of priority:
- Parameters specified on the command line (
--parameter) - Parameters that are provided using the
-params-fileoption - Config files that are provided using the
-coption - The config file named
nextflow.configin the current directory - The config file named
nextflow.configin the pipeline project directory - The config file
$HOME/.nextflow/config - Values defined within the pipeline script itself (e.g.,
main.nf)
Warning
nf-core pipeline parameters must be passed via the command line (--<parameter>) or Nextflow -params-file option. Custom config files, including those provided by the -c option, can be used to provide any configuration except for parameters.
Notably, while some of these files are already included in the nf-core pipeline repository (e.g., the nextflow.config file in the nf-core pipeline repository), some are automatically identified on your local system (e.g., the nextflow.config in the launch directory), and others are only included if they are specified using run options (e.g., -params-file, and -c).
Understanding how and when these files are interpreted by Nextflow is critical for the accurate configuration of a pipeline's execution.
Parameters¶
Parameters are pipeline specific settings that can be used to customise the execution of a pipeline.
Every nf-core pipeline has a full list of parameters on the nf-core website (for example here are the nf-core demo pipeline parameters). When viewing these parameters online, you will also be shown a description and the type of the parameter. Some parameters will have additional text to help you understand when and how a parameter should be used.
Parameters and their descriptions can also be viewed in the command line using the run command with the --help parameter:
Exercise
View the parameters for the nf-core/demo pipeline using the command line:
Solution
The nf-core/demo pipeline parameters can be printed using the run command and the --help option:
Revision flag
Including the -r flag with a specific revision ensures that you are always running the same version of the pipeline, which is important for reproducibility.
Parameters in the command line¶
At the highest level, parameters can be customised using the command line. Any parameter can be configured on the command line by prefixing the parameter name with a double dash (--):
When to use -- and -
Nextflow options are prefixed with a single dash (-) and pipeline parameters are prefixed with a double dash (--).
Depending on the parameter type, you may be required to add additional information after your parameter flag. For example, for a string parameter, you would add the string after the parameter flag:
Exercise
Give the MultiQC report for the nf-core/demo pipeline the name of your favourite animal using the multiqc_title parameter using a command line flag:
Solution
Add the --multiqc_title flag to your command and execute it. Use the -resume option to save time:
nextflow run nf-core/demo -profile test,apptainer -r 1.1.0 --outdir results --multiqc_title kiwi -resume
In this example, you can check your parameter has been applied by listing the files created in the results folder (results):
--multiqc_title is a parameter that directly impacts a result file. For parameters that are not as obvious, you may need to check your log to ensure your changes have been applied. You should not rely on the changes to parameters printed to the command line when you execute your run:
Default configuration files¶
All parameters will have a default setting that is defined using the nextflow.config file in the pipeline project directory. By default, most parameters are set to null or false and are only activated by a profile or configuration file.
There are also several includeConfig statements in the nextflow.config file that are used to include additional .config files from the conf/ folder. Each additional .config file contains categorised configuration information for your pipeline execution, some of which can be optionally included:
base.config- Included by the pipeline by default.
- Generous resource allocations using labels.
- Does not specify any method for software management and expects software to be available (or specified elsewhere).
igenomes.config- Included by the pipeline by default.
- Default configuration to access reference files stored on AWS iGenomes.
modules.config- Included by the pipeline by default.
- Module-specific configuration options (both mandatory and optional).
test.config- Only included if specified as a profile.
- A configuration profile to test the pipeline with a small test dataset.
test_full.config- Only included if specified as a profile.
- A configuration profile to test the pipeline with a full-size test dataset.
Notably, configuration files can also contain the definition of one or more profiles. A profile is a set of configuration attributes that can be activated when launching a pipeline by using the -profile command option:
Profiles used by nf-core pipelines include:
- Software management profiles
- Profiles for the management of software using software management tools, e.g.,
docker,apptainer, andconda.
- Profiles for the management of software using software management tools, e.g.,
- Test profiles
- Profiles to execute the pipeline with a standardised set of test data and parameters, e.g.,
testandtest_full.
- Profiles to execute the pipeline with a standardised set of test data and parameters, e.g.,
Multiple profiles can be specified in a comma-separated (,) list when you execute your command. The order of profiles is important as they will be read from left to right:
nf-core pipelines are required to define software containers and conda environments that can be activated using profiles. Although it is possible to run the pipelines with software installed by other methods (e.g., environment modules or manual installation), using software containers is more convenient and more reproducible.
Tip
If your computer has internet access and one of Conda, Apptainer, or Docker installed, you should be able to run any nf-core pipeline with the test profile and the respective software management profile 'out of the box'.
The test data profile will pull small test files directly from the nf-core/test-data GitHub repository and run it on your local system. The test profile is an important control to check the pipeline is working as expected and is a great way to trial a pipeline. Some pipelines have multiple test profiles for you to try.
Shared configuration files¶
An includeConfig statement in the nextflow.config file is also used to include custom institutional profiles that have been submitted to the nf-core config repository. At run time, nf-core pipelines will fetch these configuration profiles from the nf-core config repository and make them available.
For shared resources such as an HPC cluster, you may consider developing a shared institutional profile. You can follow this tutorial for more help.
Custom configuration files¶
Nextflow will also look for files that are external to the pipeline project directory. These files include:
- The config file
$HOME/.nextflow/config - A config file named
nextflow.configin your current directory - Custom files specified using the command line
- A parameter file that is provided using the
-params-fileoption - A config file that are provided using the
-coption
- A parameter file that is provided using the
You don't need to use all of these files to execute your pipeline.
Parameter files
Parameter files are .json files that can contain an unlimited number of parameters:
{
"<parameter1_name>": 1,
"<parameter2_name>": "<string>",
"<parameter3_name>": true
}
You can override default parameters by creating a custom .json file and passing it as a command-line argument using the -params-file option.
nextflow run nf-core/<workflow> -profile test,apptainer -r 1.1.0 --outdir results -params-file <path/to/params.json>
Exercise
Give the MultiQC report for the nf-core/demo pipeline the name of your favourite food using the multiqc_title parameter in a parameters file:
Solution
Create a custom .json file that contains your favourite food, e.g., cheese:
Include the custom .json file in your execution command with the -params-file option:
nextflow run nf-core/demo -profile test,apptainer -r 1.1.0 --outdir results -params-file my-custom-params.json
Check that it has been applied:
Configuration files
Configuration files are .config files that can contain various pipeline properties. Custom paths are passed on the command line using the -c option:
Multiple custom .config files can be included at execution by separating them with a comma (,).
Custom configuration files follow the same structure as the configuration file included in the pipeline directory.
Configuration properties are organised into scopes by dot prefixing the property names with a scope identifier or grouping the properties in the same scope using the curly brackets notation. For example:
Is equivalent to:
Scopes allow you to quickly configure settings required to deploy a pipeline on different infrastructure using different software management. For example, the executor scope can be used to provide settings for the deployment of a pipeline on a HPC cluster. Similarly, the apptainer scope controls how Apptainer containers are executed by Nextflow.
Multiple scopes can be included in the same .config file using a mix of dot prefixes and curly brackets. A full list of scopes is described in detail in the documentation.
Exercise
Give the MultiQC report for the nf-core/demo pipeline the name of your favourite colour using the multiqc_title parameter in a custom .config file:
Solution
Create a custom .config file that contains your favourite colour, e.g., blue:
Include the custom .config file in your execution command with the -c option:
nextflow run nf-core/demo -profile test,apptainer -r 1.1.0 --outdir results -resume -c custom.config
Check that it has been applied:
Why did this fail?
You can not use the params scope in custom configuration files. Parameters can only be configured using the -params-file option and the command line. While the parameter is listed in stdout, it was not applied to the executed command:
The process scope allows you to configure pipeline processes and is used extensively to define resources and additional arguments for modules.
By default, process resources are allocated in the conf/base.config file using the withLabel selector:
Similarly, the withName selector enables the configuration of a process by name. By default, module parameters are defined in the conf/modules.config file:
While some tool arguments are included as a part of a module. To make modules sharable across pipelines, most tool arguments are defined in the conf/modules.config file in the pipeline code under the ext.args entry.
Importantly, having these arguments outside of the module also allows them to be customised at runtime.
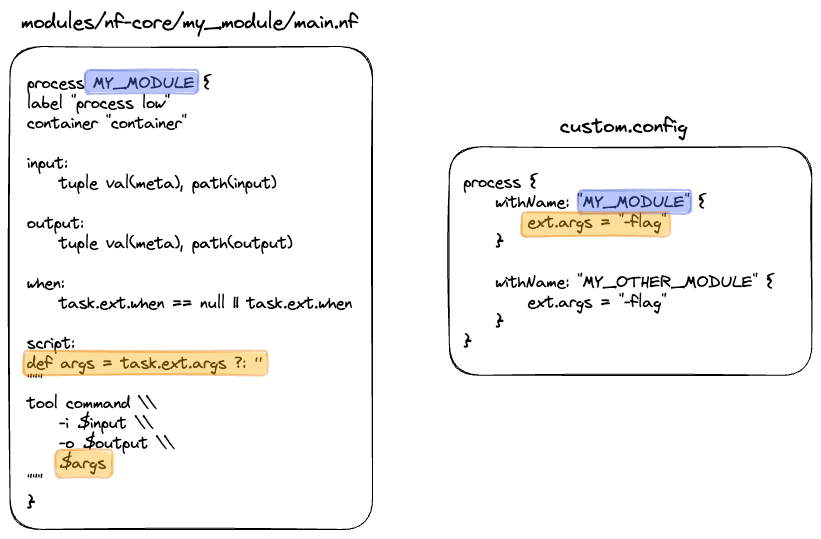
For example, if you were trying to add arguments in the MULTIQC process in the nf-core/demo pipeline, you could use the process scope:
However, if a process is used multiple times in the same pipeline, an extended execution path of the module may be required to make it more specific:
process {
withName: "NFCORE_DEMO:DEMO:MULTIQC" {
ext.args = "<your custom parameter>"
}
}
The extended execution path is built from the pipelines, subworkflows, and modules used to execute the process.
In the example above, the nf-core MULTIQC module was called by the DEMO workflow, which was called by the NFCORE_DEMO pipeline in the main.nf file.
How to build an extended execution path
It can be tricky to evaluate the path used to execute a module. If you are unsure of how to build the path you can copy it from the conf/modules.config file. How arguments are added to a process can also vary. Be vigilant.
Exercise
Create a new .config file that uses the process scope to overwrite the args for the MULTIQC process. Change the args to your favourite month of the year, e.g, "--title \"october\"".
In this example, the \ is used to escape the " in the string. This is required to ensure the string is passed correctly to the MULTIQC module.
Solution
Make a custom config file that uses the process scope to replace the args for the MULTIQC process:
Execute your run command again with the custom configuration file:
nextflow run nf-core/demo -r 1.1.0 -profile test,apptainer --outdir results -resume -c custom.config
Check that it has been applied:
Exercise
Demonstrate the configuration hierarchy using the nf-core/demo pipeline by adding a params file (-params-file), and a command line flag (--multiqc_title) to your execution. You can use the files you have already created. Make sure that the --multiqc_title is different to the multiqc_title in your params file and different to the title you have used in the past.
Solution
Use the .json file you created previously:
Execute your command with your params file (-params-file) and a command line flag (--multiqc_title):
nextflow run nf-core/demo -r 1.1.0 -profile test,apptainer --outdir results -resume -params-file my-custom-params.json --multiqc_title "cake"
In this example, as the command line is at the top of the hierarchy, the multiqc_title will be "cake".
Key points
- nf-core pipelines follow a similar structure
- nf-core pipelines are configured using multiple configuration sources
- Configuration sources are ranked to decide which settings to apply
- Pipeline parameters must be passed via the command line (
--<parameter>) or Nextflow-params-fileoption