ggtree v3.14.0 Learn more at https://yulab-smu.top/contribution-tree-data/
Please cite:
Guangchuang Yu. Using ggtree to visualize data on tree-like structures.
Current Protocols in Bioinformatics. 2020, 69:e96. doi:10.1002/cpbi.96
library(phangorn)
Loading required package: ape
Attaching package: 'ape'
The following object is masked from 'package:ggtree':
rotate
library(ggplot2)library(ggtreeExtra)
ggtreeExtra v1.16.0 For help: https://yulab-smu.top/treedata-book/
If you use the ggtree package suite in published research, please cite
the appropriate paper(s):
S Xu, Z Dai, P Guo, X Fu, S Liu, L Zhou, W Tang, T Feng, M Chen, L
Zhan, T Wu, E Hu, Y Jiang, X Bo, G Yu. ggtreeExtra: Compact
visualization of richly annotated phylogenetic data. Molecular Biology
and Evolution. 2021, 38(9):4039-4042. doi: 10.1093/molbev/msab166
library(ape)library(dplyr)
Attaching package: 'dplyr'
The following object is masked from 'package:ape':
where
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
library(tidyr)
Attaching package: 'tidyr'
The following object is masked from 'package:ggtree':
expand
Import Newick tree file generated by FastTree and created a tree object
tree <-read.tree("~/Desktop/GA2025/01.Short_read_workshop/SR_workshop/temp/tree_file.txt")
Now we can visualize the phylogenetic tree with ggtree
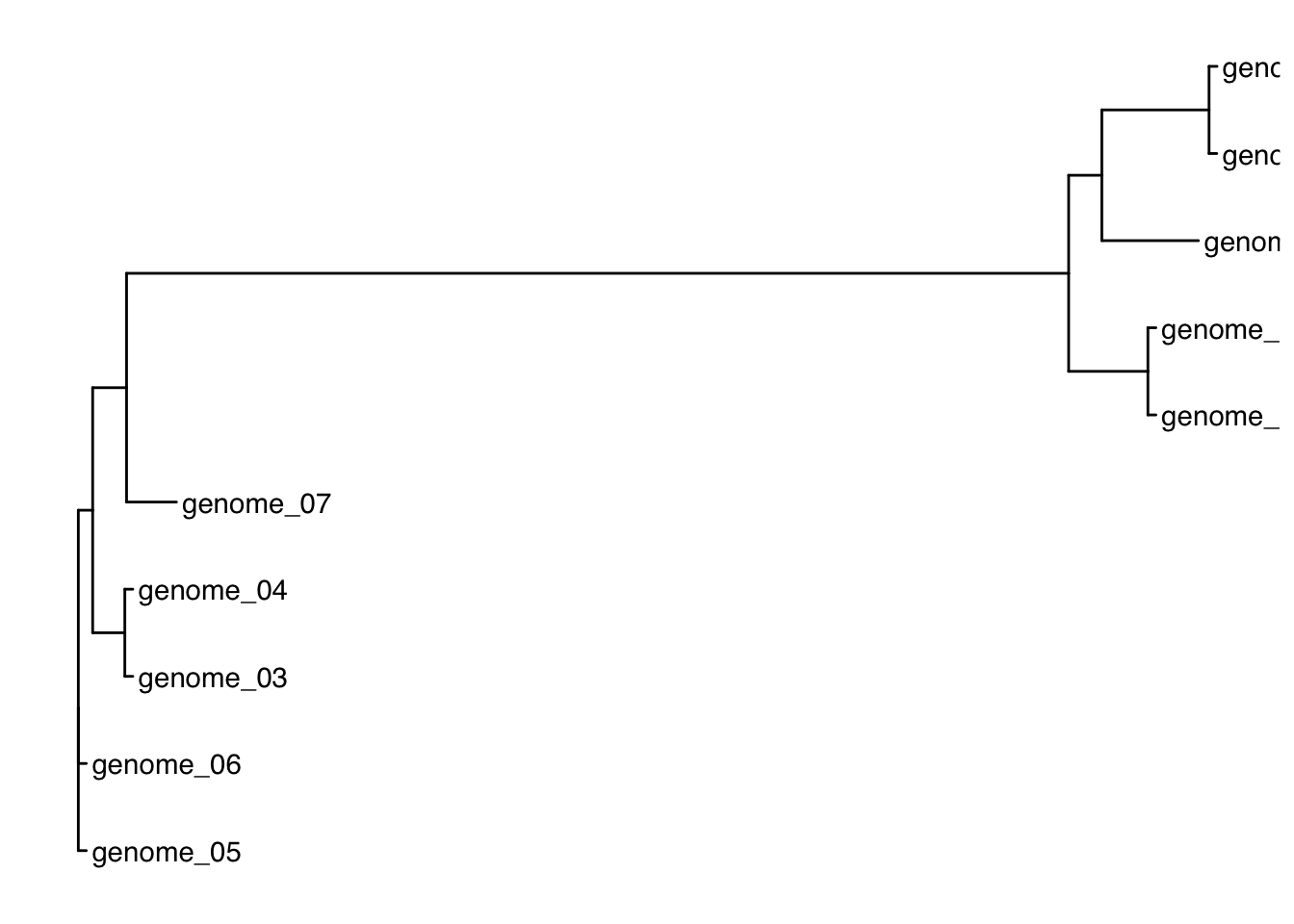
ggtree(tree) +geom_tiplab()
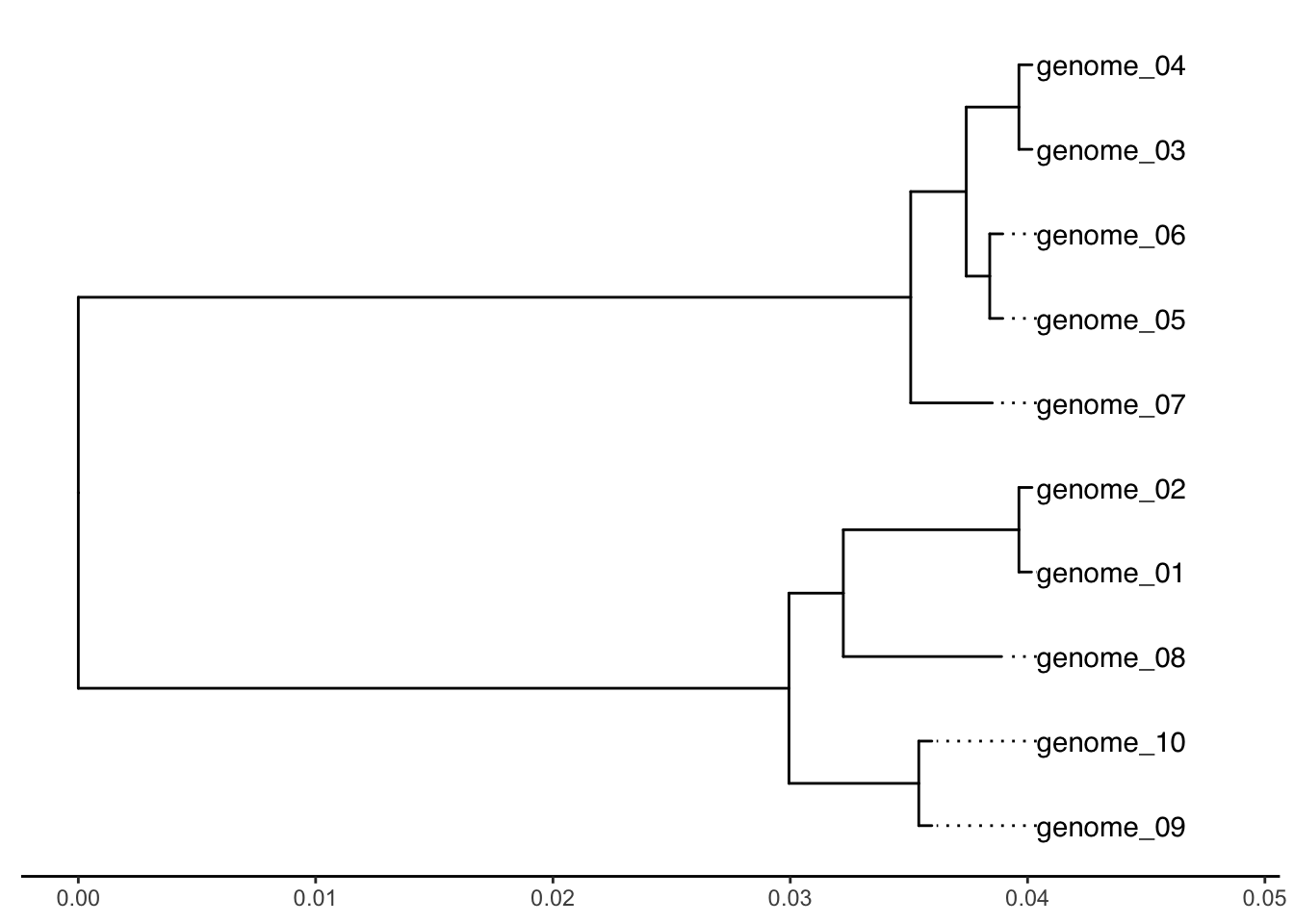
This quick and easy tree doesn’t include a reference genome, so we can use the midpoint() function of the phangorn package to reroot the tree at midpoint (i.e. locate the midpoint of the longest path between any two tips and place the root at that location, assuming a constant evolutionary rate).
#Rerooting tree at midpointreroot_tree <-midpoint(tree)# Rescale tree based on the maximum tree depth and plot a cleaned up version of the treetree_depth <-max(node.depth.edgelength(reroot_tree))p0 <-ggtree(reroot_tree) +geom_tiplab(align =TRUE, linesize =0.5) +xlim(0, tree_depth *1.2) +theme_tree2()p0
Let’s now visualise some of the genome quality metrics that were gathered during the workshop. Read in the genome metadata.
qual <-read.delim("~/Desktop/GA2025/01.Short_read_workshop/SR_workshop/temp/genome_quality.txt")
Join tree info with metadata.
# Generate random values for each tip label in the datad1 <-data.frame(id=reroot_tree$tip.label)colnames(d1)[1] <-"ID"d1 <-left_join(d1, qual, "ID")
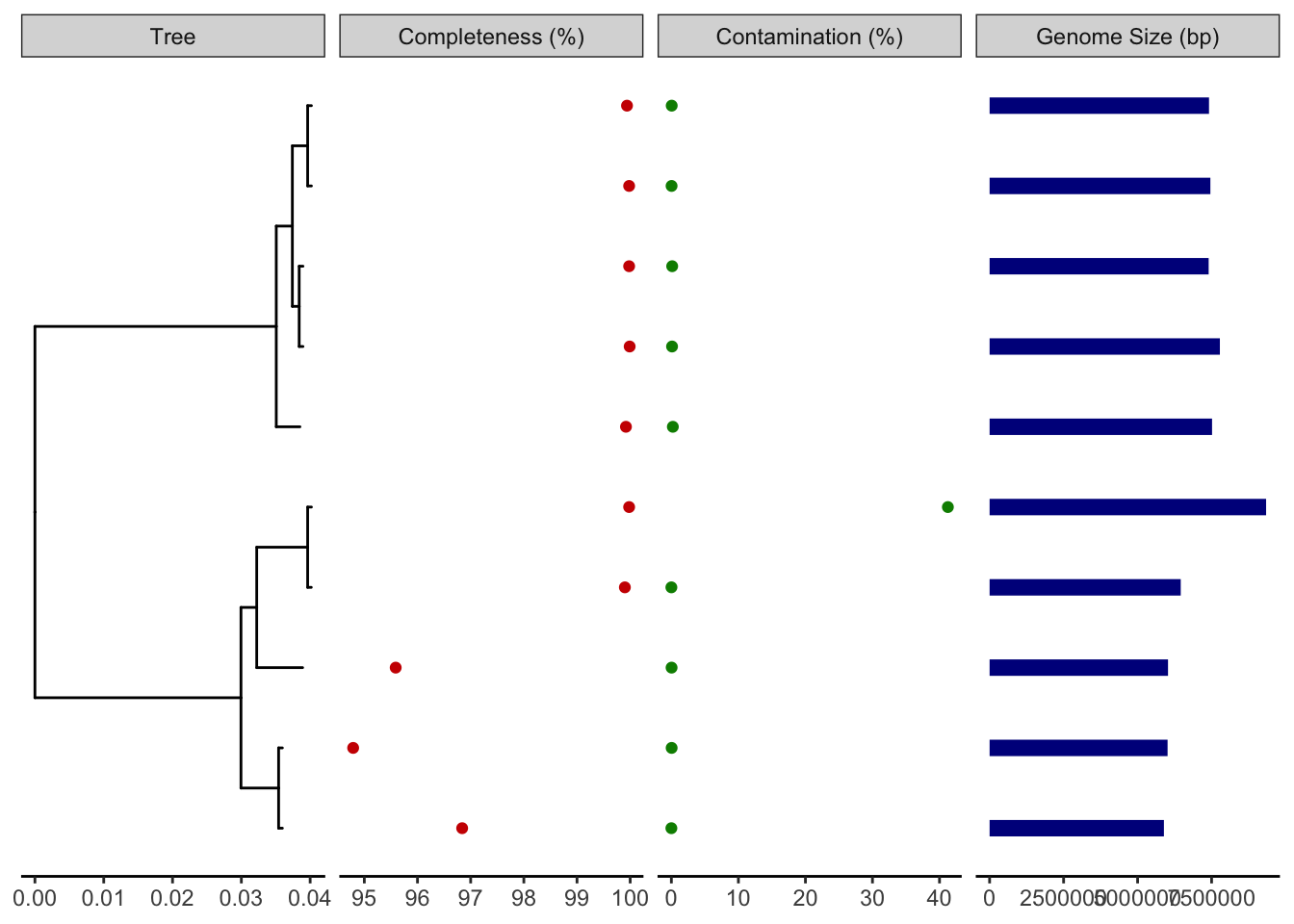
Facet plot for completeness, contamination, and genome size.
#Facet for treep1 <-ggtree(reroot_tree)#Facet for genome completenessp2 <-facet_plot(p1, panel="Completeness (%)", data=d1, geom=geom_point, aes(x=Completeness), color='red3')#Facet for genome contaminationp3 <-facet_plot(p2, panel="Contamination (%)", data=d1, geom=geom_point, aes(x=Contamination), color='green4')#Facet for genome sizep4 <-facet_plot(p3, panel="Genome Size (bp)", data=d1, geom=geom_segment, aes(x=0, xend=Genome_Size, y=y, yend=y), size=3, color='blue4')# Show all three plots with a scalep4 +theme_tree2()