Preparing for sequencing
You’ve done your experiment, extracted some DNA or RNA, and want to sequence it, but now what?
Quality control of DNA/RNA
Your DNA or RNA samples will need to pass certain quality cut-offs for sequencing, which the sequencing facility will ask for and will have thresholds they require upon submission (e.g., see the Otago Genomics Facility requirements here). After you have done your extractions in the lab, you should perform QC with:
- Agarose gel electrophoresis. For RNA, this will give you a good indication of the integrity of your RNA sample; generally you will expect to see two clear bands for the 28S and 18S rRNA subunits. For DNA samples, in particular for long-read sequencing, you want to see a high molecular weight band with minimal smearing (which would indicate degradation). Getting the high-molecular-weight gDNA (i.e., long gDNA fragments) is critical to obtaining high-quality data for WGS using ONT/PacBio.
Making and performing agarose gel electrophoresis is a core molecular biology skill that we expect almost all readers will have learnt, so we will not go into further details here. Note for RNA samples, different species can have different rRNA banding patterns that may not look like a ‘classic’ 28S/18S pattern (for eukaryotes), and different tissue types can have different levels of small RNAs, which can appear like degraded RNA on a gel. You may need to do some literature searching if you have odd results. For high molecular weight DNA (i.e., for genome sequencing), you may need to run a special gel e.g., a pulsed-field gel electrophoresis or a capillary-based system such as Agilent Femto Pulse System. See also assessing molecular weight in Nanopore documentation.
- A microvolume spectrophotometer (e.g., NanoDrop, DeNovix), which will give you a good indication of the purity/contamination of your sample through 260/280 and 260/230 ratios. This method is not very reliable for determining concentration of your nucleic acid.
There is usually a NanoDrop or DeNovix in most labs as standard benchtop equipment (NanoDrops are more common and most people refer to this QC method simply as ‘nanodrop’). It is free to run (no additional reagents required beyond UltraPure water/ddH2O) and only requires 1-2 μL of your sample. Results appear on the screen within seconds. Gently clean with UltraPure water and a kimwipe between samples; do not use ethanol on platform. While the nanodrop is not a great option for definitive quantification of your sample concentration, it can be a good starting indication as it can measure a broader concentration range than the Qubit. It will almost always overestimate the concentration. If your Qubit to nanodrop concentration ratio is more than 50% different (e.g., nanodrop says 100 ng/μL, Qubit says <50ng/μL), this could indicate substantial contamination of the undesired nucleic acid or other contaminant and you may need to re-purify / clean and concentrate samples.
- A fluorescent-based quantification method (e.g., Qubit), which will give you a very accurate indication of concentration of your nucleic acid (but not of any contaminants).
Many labs have a Qubit (e.g., see Qubit 4 model; other models exist), but they are less common than microvolume spectrophotometers. You may need to ask around your department/faculty to see where one is and who owns it. There are a few ongoing costs associated with using the Qubit (standards, buffers, dyes and tubes are required consumables), so if you are borrowing this from another group you will need to work out how the costs should be covered. The estimated cost is around $1 per sample (adds up fast when you have many samples + technical replication!). You will also have to determine which kit you need, often either the dsDNA HS (high sensitivity) assay kit or the RNA HS kit; there are also broad range kits, see assay types here. The Qubit generally requires 1 μL of your sample to run. Samples for the Qubit take a few mins to prep, then results are read within seconds and displayed on the screen. DNA/RNA concentrations can both be measured very accurately, but not simultaneously. The Qubit is very sensitive, but in a more narrow concentration range to the NanoDrop. You may need to use the NanoDrop concentration as a general indication for diluting your samples to the concentration range of the Qubit kit you are using, so read the kit information to determine which one you need.
- A fragment analysis instrument (e.g., Agilent 2100 Bioanalyzer, Agilent 5300 Fragment Analyzer or Agilent TapeStation), which you can think of like a high-tech agarose gel. It will give you a good indication of your 28S/18S ratio for RNA (this is used to determine the overall RIN - RNA integrity number*) or your fragment size distribution for DNA.
You will most likely find one of these instruments as part of a service offered by a genetics/genomics facility within your University or Institute. They are not part of the standard benchtop lab equipment, as they require training to use and have ongoing maintenance. You should run Qubit first to get the concentration of your sample, then generally you will submit around 2-3 μL to a technician who will run the fragment analysis for you (they may need to dilute the sample and will ask for the concentration).
All three instruments above essentially function the same, but have different throughput capacity. The cost to running a sample is more than Qubit and NanoDrop; e.g., for one Bioanalyzer run, which can take up to 11 samples, this is around NZD$100 in consumables (one chip). The Fragment Analyzer and TapeStation have higher throughput, which can make it cheaper per sample if you have many, or the technician may plan to run your samples at the same time as other people’s samples on one multiwell plate.
The run will take approximately 1 hour, and results will then be available immediately (the technician will likely email a pdf file to you). You can read the electropherogram in a similar way to an agarose gel; there are many guides online to help you e.g., see here Interpretation of Bioanalyzer Traces from the University of Rochester Genomics Research Center. Note the results will likely include an estimated concentration value - it is best to not use this value going in to library prep. Stick to the Qubit results.
Lastly, the DV200 value can also be used for FFPE/degraded samples, see Agilent documentation here.
Together, these will give you an almost complete picture of your sample, ready for sequencing. Note that if your samples are below threshold quality, there are library prep protocols that can allow for lower quality or degraded samples. Ideally, this should only be done if there is no option to re-extract, re-purify or repeat the experiment.
*RNA integrity number: note that for some protosome species (e.g., molluscs, arthropods) the 28S rRNA subunit has a hidden break, which can negatively affect the RIN calculation. This results in the quality of the RNA appearing to be is worse than it actually is.
Handy tip! It is best to run nanodrop and qubit immediately after sample extraction, to avoid freeze-thaw cycles. Put a small aliquot (2-3 μL) of your sample aside too, ready to submit for fragment analysis.
Sequencing libraries
The first thing that needs to be done for Illumina, PacBio or Oxford Nanopore (ONT) sequencing is to turn the DNA or RNA sample into something called a library. This converts the raw nucleic acids into a form that the sequencer can actually read. This is generally done by the technician who will also sequence your samples, but some labs may do library prep in-house (i.e., you may do it yourself!). For RNA samples, the Illumina and PacBio platforms require that the RNA is first converted into DNA during the library prep process. ONT has the added advantage that you can sequence native RNA directly, or you can convert it to cDNA for sequencing. DNA library preparation for ONT sequencing is also generally simpler than for Illumina and PacBio. This is because the technology used to sequence the DNA/RNA using ONT is quite different to how Illumina and PacBio achieve it. There are pros and cons to all three technologies–more on that in the next section on flow cells and sequencing platforms 101.
Handy note: You can think of one library as being equivalent to one sample. It is possible to make multiple libraries from a single sample, but it is not often done this way.
Library preparation generally involves the following steps, and takes 1-2 days in the lab for Illumina and PacBio, and half a day to 1 day for ONT:
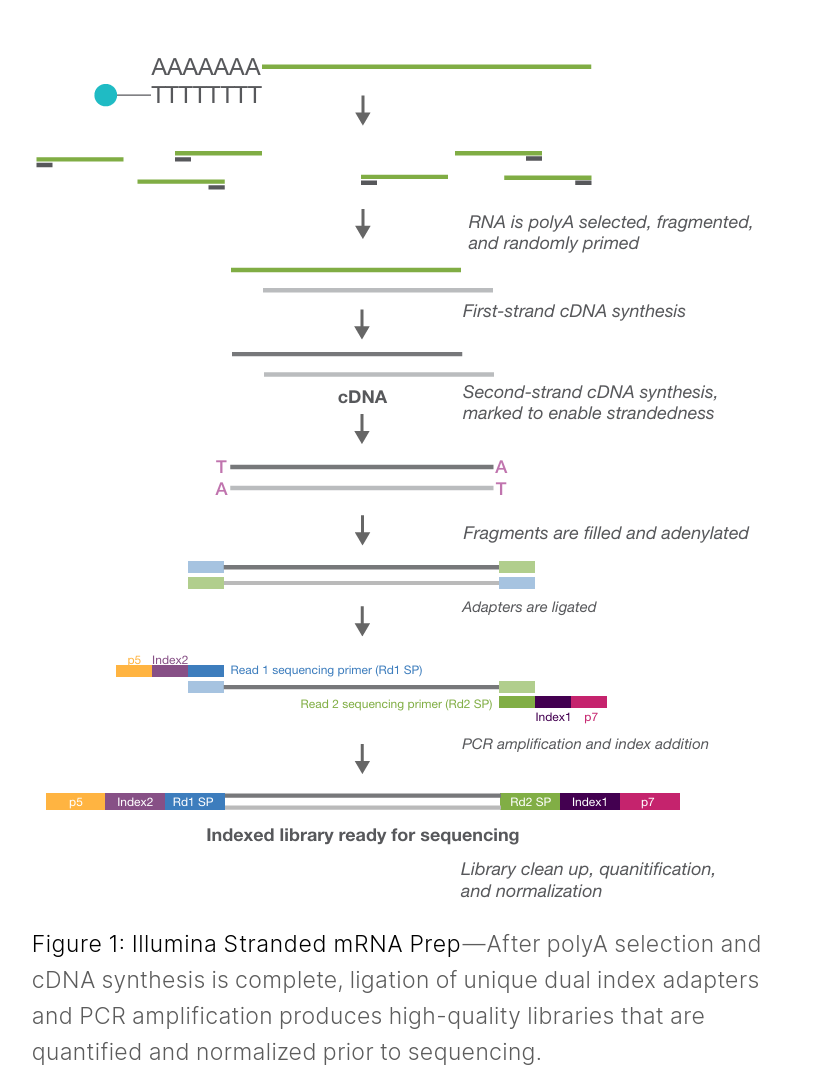
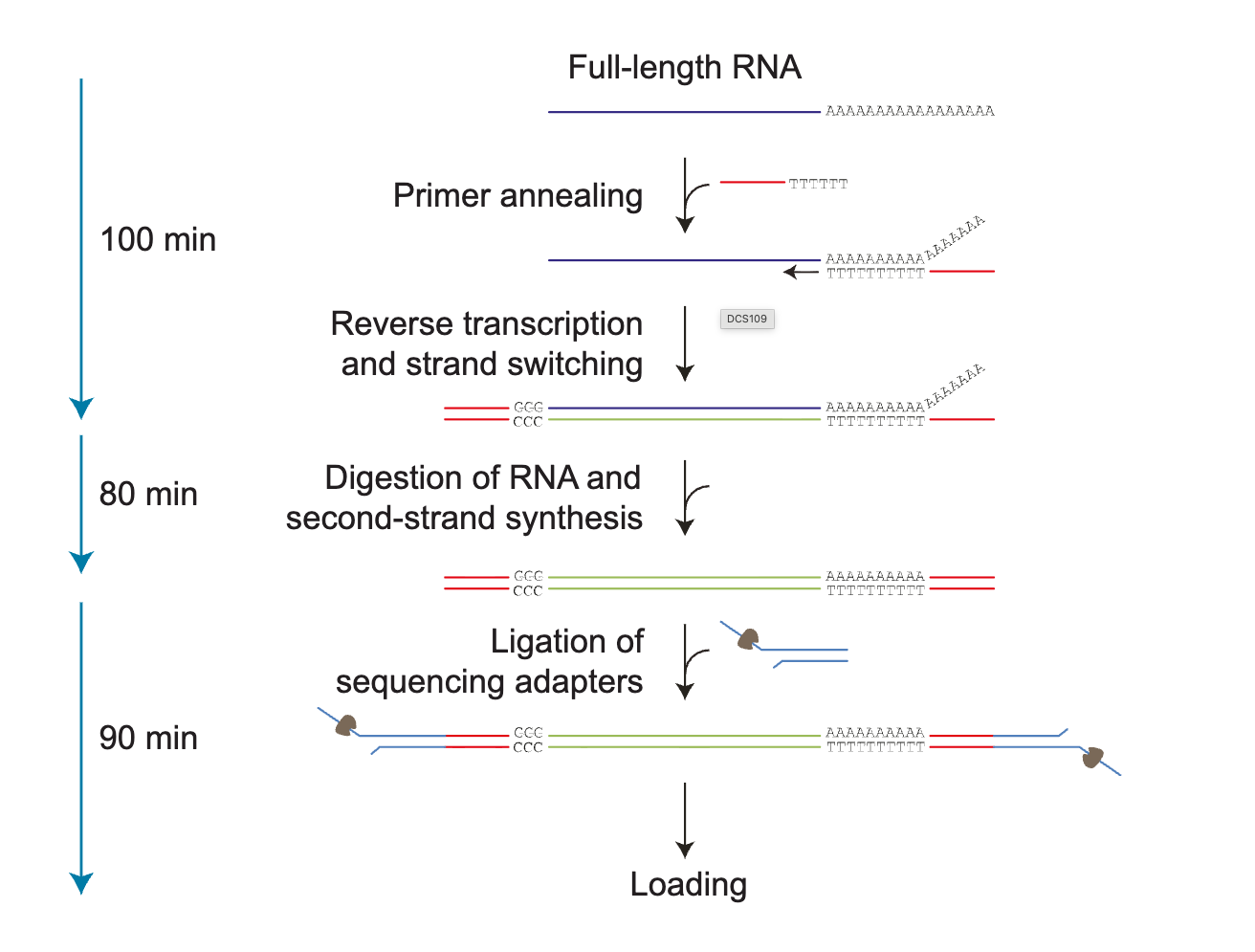
Image from link here.
(Optional) Target enrichment / molecule selection
Selectively enriching for your target molecule (e.g., polyA capture for standard RNAseq; exome or targeted gene panels in DNA sequencing). The timing of this step depends on the platform and molecule type.(RNAseq only) Converting your RNA into cDNA using reverse transcriptase. ONT also allows for direct RNA sequencing; see note below.
Fragmentation (if needed) Some kits fragment RNA before reverse transcription, others fragment cDNA/DNA after.
This ensures fragments are the right size for the platform (e.g., ~200–400 bp short reads for Illumina). Long-read platforms (ONT and PacBio) generally do not require fragmentation, but some size selection or shearing may be performed.End repair and A-tailing
Ends are cleaned up so adapters can be ligated efficiently.Adapter ligation
Adapters are short sequences that enable library binding to the flow cell (Illumina or ONT) or capture for SMRTbells (PacBio).
Some kits combine this with indexing.
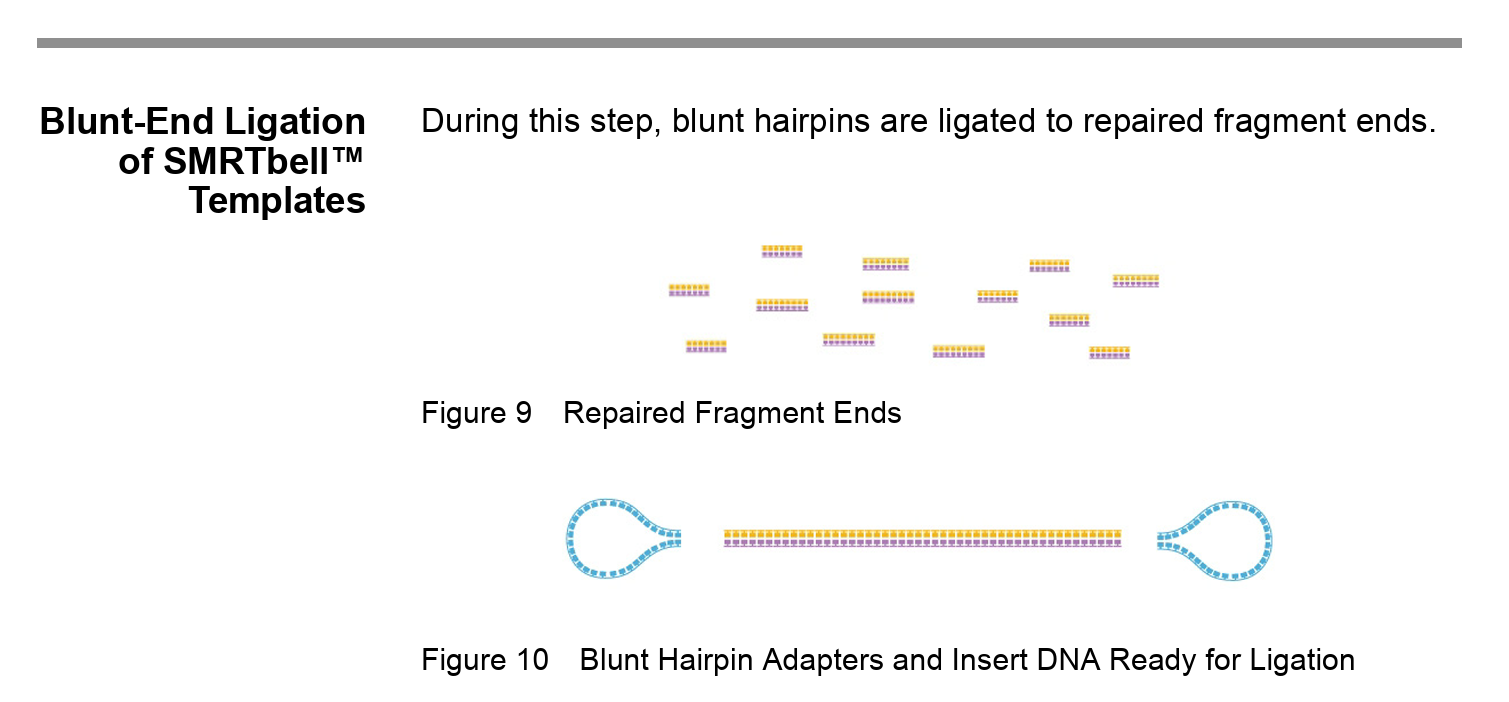
Note: the SMRTbell technology allows HiFi (high fidelity) long read sequencing. The DNA becomes circularised, which allows the polymerase to make repeated passes around the DNA and the consensus sequence therefore has a higher accuracy than single pass sequencing.
Indexing (barcoding)
Indices allow multiple samples to be pooled together and sequenced on the same flow cell, then computationally (in silico) separated afterward (called mulitplexing and demultiplexing).Size selection / cleanup
Typically done with magnetic beads to remove adapter dimers and select the desired fragment range.Library amplification (if required)
Some protocols use PCR to enrich adapter-ligated molecules; others (e.g., some PacBio) are PCR-free. ONT has a cDNA-PCR sequencing protocol for lower input RNA samples.Final QC and quantification
Using Qubit, Bioanalyzer/Tapestation/Fragment Analyzer, etc. This step ensures your library meets sequencing requirements.
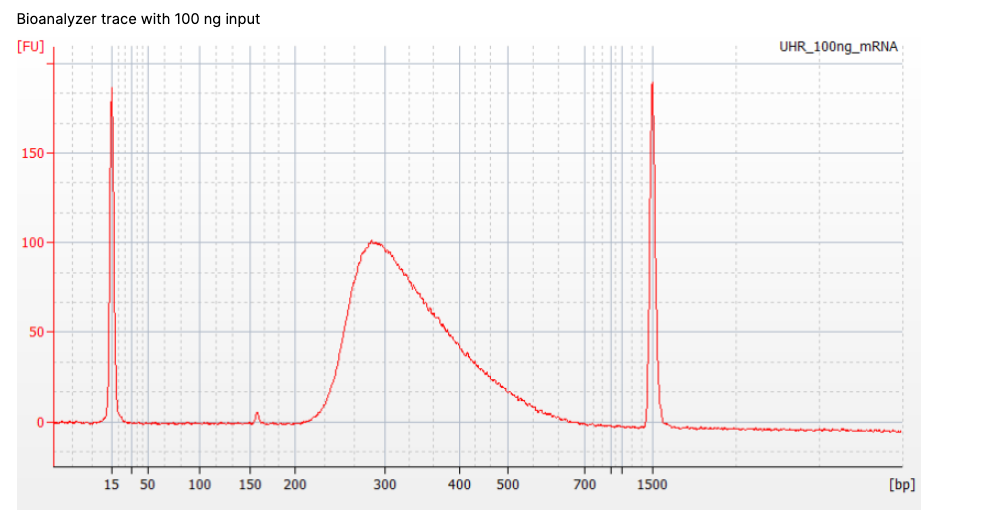
Q: What do you think the two large narrow peaks are around 15 bp and 1500bp?
The two narrow peaks are ladder sequence. These are internal standards of known size we add in for quality control. These can also be used for concentration estimation, but fluorescence-based quantification (e.g., Qubit) is more accurate.
Direct RNA sequencing with ONT: Native RNA can be sequenced directly with ONT, which allows exploring of modified bases (e.g., methylated bases). It takes approximately 140 mins to complete library preparation. Currently, there is no option for multiplexing, although a multiplexing kit is scheduled for release in 2026.
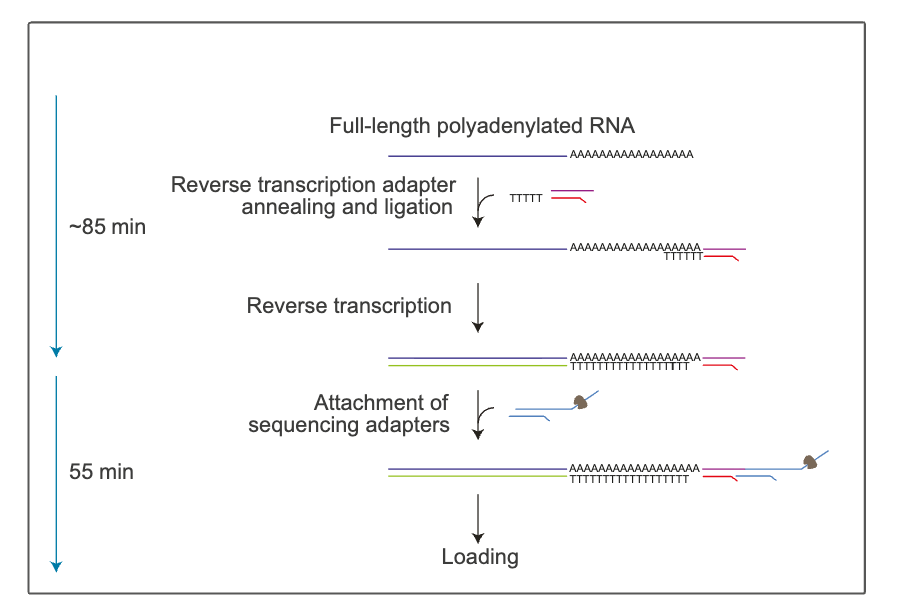
Note that the second complementary cDNA strand is synthesised for stability by reverse transcription. The cDNA strand is not sequenced, but improves the RNA sequencing output.
There are different library prep methods (i.e., protocols) for each platform that you will need to chose. This will depend on a few things, such as:
- The quality of your RNA/DNA (e.g., high-quality RNA allows polyA selection; degraded samples may require ribo-depletion or specialised kits).
- The species/tissue type you extracted your RNA/DNA from (e.g., plants have rRNA types that require plant-specific depletion kits, some tissues have high mitochondrial RNA content).
- The type of analysis you want to do i.e., what is your research question.
For example, if you are doing a ‘generic’ RNA sequencing project (e.g., you plan to do differential gene expression analysis to compare different samples), a common choice is Illumina stranded mRNA library prep, which uses polyA selection to capture mRNA. However, you may chose Illumina Stranded total RNA library prep with ribo-depletion, which is more expensive, but it has some advantages such as: it can capture non-polyadenylated RNAs (more comprehensive RNA profile) and and is a better option if your samples are partially degraded (it can also handle FFPE samples).
Total RNA library prep with ribo-depletion is the only option for prokaryotic RNA sequencing, as prokaryotes do not have polyA tails.
There are two main versions of the Illumina RNA library prep kit chemistry. The “Illumina TruSeq” kit uses an older chemistry and the “Illumina Stranded” kit uses a newer chemistry. Within each of these versions are also several types of kits for specific purposes (e.g,. total RNA or mRNA). Both chemistries are very robust options and produce good quality sequence data. The TruSeq chemistry requires a slightly higher minimum mRNA input (100 ng) vs. the minimum mRNA input for Illumina Stranded (25 ng), so if you have very low amounts of RNA you may need to use the “Illumina Stranded” kit. The newer “Illumina Stranded” also has the advantages of being a slightly faster protocol to complete, and allows for higher multiplexing (up to 384 samples, vs up to 96 samples for TruSeq). Note that both chemistries do strand specific sequencing. For most generic RNAseq projects, it won’t matter which one you use. You may note that some NZ sequencing facilities only offer one or the other chemistry.
See here for comparison between Illumina TruSeq stranded and Illumina stranded mRNA kits.
DISCUSSION 💬
Mature mRNAs have polyA tails, which can be selectively isolated using oligo DT coated beads that bind mature RNAs only. All other non-polyadenylated nucleic acids and cellular debris can then be washed away.
Indexing (barcoding) allows multiple samples to be pooled in a single sequencing run. This massively reduces cost and time. Because each library carries a unique index, they can be mixed (i.e., pooled) together and sequenced simultaneously, and downstream software can computationally separate (demultiplex) them accurately afterward.
Size selection ensures that fragments fall within the size range the sequencer expects. Without it, you may get:
leftover adapter dimers (which waste sequencing reads as they take up ‘real estate’ on the flow cell)
too-short fragments (which cluster preferentially, causing over-representation and distorts the data e.g., sequencing may read into adapters or flow cell)
too-long fragments (which may not fully sequence or reduce yield)
The final fragment analysis (e.g., Bioanalyzer) trace of the library will show you. See below a picture of a trace with adapter dimers present (~120-150bp peak).
Tip: You can repeat the final wash step in the library prep protocol to remove adapter dimers and re-run Bioanalyzer to confirm. Sometimes a very small peak will still be present which will not overly affect the run (you can see one on the previous image above!)
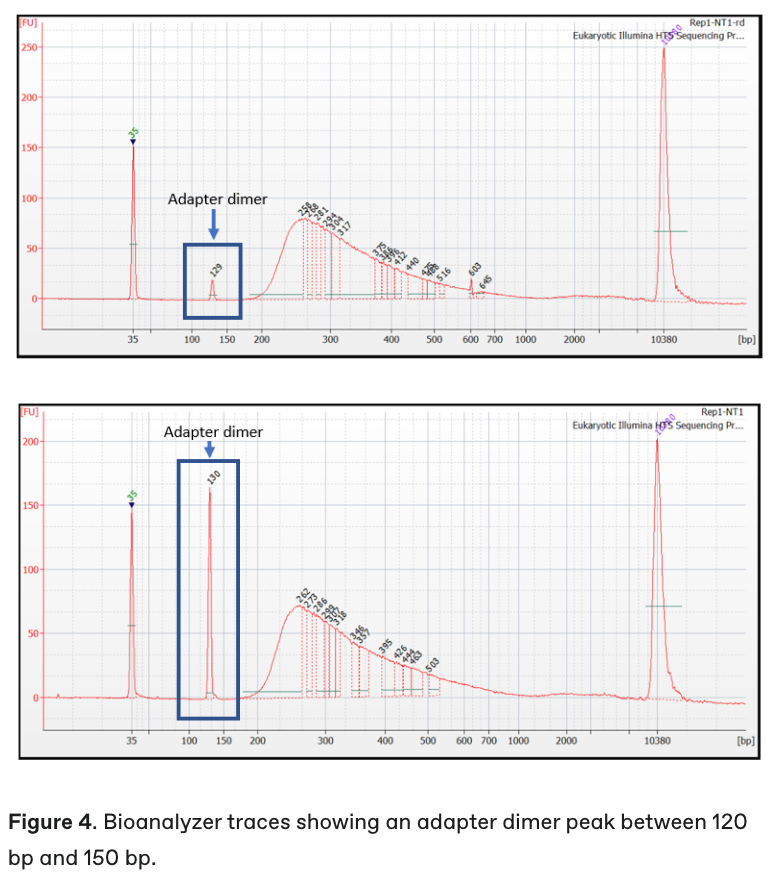 Image from Illumina general knowledge base
Image from Illumina general knowledge base
Further reading 📚
Auckland Genomics provide great documention on choosing an approach for your sequencing project.
DNA sequencing: whole genome, shotgun or metagenomic; which one is right for you?
RNA sequencing: bulk RNA-seq, single cell RNA-seq, mRNA vs total, ONT RNA-seq; which one is right for you?
Flow cells and sequencing platforms 101
The three major sequencing companies are Illumina, Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), which each produce their own platforms (i.e., the instruments or machines that perform the sequencing) and consumables (i.e., the flow cells and reagents). Each make different platforms that can handle different levels of throughput, but the chemistry and the ‘reading’ of the sequencing is the main point of difference between the three companies.
A few examples of the different platforms are (throughput in brackets):
Illumina
- MiSeq i100 (small)
- NextSeq 550 / 1000 / 2000 (medium)
- NovaSeq 6000 / X (large)
PacBio
- Sequel II
- Revio
Oxford Nanopore Technologies
- MinION (small)
- GridION (medium)
- PromethION 2 Solo / 2 Integrated / 24 (large - very large)
A flow cell is the physical surface inside the sequencing machine (or platform) where the actual reading of DNA or RNA occurs. There are different sizes you can chose from, depending on how many reads you need. Although Illumina, PacBio, and ONT all call their consumables “flow cells,” the underlying technologies are very different.
For the most part, and in-depth knowledge of how these flow cells work is not needed to get you started with your sequencing project. The more important thing to understand is the strengths and limitations of the different technologies and how to chose the right kind of sequencing for your research project.
Here are the major differences between the technologies:
| Feature | Illumina | PacBio | Oxford Nanopore (ONT) |
|---|---|---|---|
| How sequencing works | Sequencing-by-synthesis (fluorescent nucleotides added one base at a time) | Single-molecule real-time (SMRT) sequencing (polymerase incorporates fluorescent bases inside Zero-Mode Waveguides (ZMW)) | Nanopore sensing (changes in ionic current as DNA/RNA passes through a pore) |
| Flow cell structure | Patterned flow cell with billions of oligonucelotides that form clonal clusters | SMRT Cell containing millions of ZMWs | Membrane embedded with thousands of protein nanopores |
| What binds to the flow cell | Libraries bind via adapters to oligonucelotides → amplified into clusters | A single SMRTbell + polymerase complex loads into each ZMW | DNA or RNA strand with a motor protein threads into a nanopore |
| Signal detected | Fluorescent signal imaged each cycle | Fluorescent flashes when each base is incorporated | Changes in electrical current across the pore |
| Amplification? | Yes — cluster generation required | No — single-molecule sequencing. HiFi (high fidelity) reads are generated by multiple passes of the same circular molecule to create a consensus | No (PCR-free), though can use PCR in library prep |
| Typical read length | 100–300 bp | 10–25 kbp HiFi reads | 10 kbp to >100 kbp (ultra-long >1 Mbp possible). Short reads e.g., 100bp also possible. |
| Can sequence native RNA? | No — convert to cDNA library | No — convert to cDNA library | Yes — direct RNA sequencing. Recognises standard and certain modified bases e.g., pseudouridine and m6A |
| Strengths | High accuracy; high throughput; cost-efficient; chemistry highly compatible with different species/tissues | Highly accurate long reads; excellent for haplotype resolution | Long and Ultra-long reads; portable; real-time analysis; can detect base modifications; enables adaptive sampling (unique software-based target enrichment or depletion method that provides multiomic view of the genome) |
| Limitations | Short reads only | Lower throughput than Illumina; expensive | Higher raw error rate; pore lifetime limits yield and susceptible to clogging; short reads possible but less accurate and lower yield |
| Best used for | Standard RNA-seq; differential gene expression (DGE); de novo transcriptomes; high-depth short-read assays; metagenomics; error-correcting long reads | Genome assembly (chromosome-level with HiFi); full-length RNA (Iso-Seq for isoforms); structural variant detection | Field-based sequencing (e.g., rapid microbial identification); genome assembly; native and full-length RNA including modification detection (e.g., methylation); isoforms detection as well as differential expression; structural variant detection |
Decision points 🤔
Choosing a platform to do your sequencing comes down to the question you are trying to answer. See the last row in the table above ‘Best used for’ for some examples of why you might pick one platform over another!
When you get in contact with a sequencing facility, the question they will ask you is not how many samples are you sequencing, but rather, how many reads do you need? The number of reads you end up with per library will be approximately evenly distributed across all libraries, as they all get pooled together in equimolar amounts into one tube before loading on to the flow cell. The more libraries in the pool, the less reads per library. Hence, it does not matter how many samples you have (to an extent), what matters is how many reads you need in total. This will determine what size flow cell you need and which platform you will use, as different platforms have different capacity (e.g., Illumina NextSeq is a ‘medium throughput’ platform, NovaSeq is a ‘large throughput’ platform). The number of reads you need per library scales with the size of the genome and/or complexity of your transcriptome.
As a general rule of thumb, for transcriptome sequencing you will need:
Table 1: Reads per sample
| Purpose / Type | Approx reads per sample |
|---|---|
| Gene expression profiling | 5–25 million |
| Complete expression + alternative splicing | 30–60+ million |
| De novo transcriptome assembly | ~100+ million |
You may be thinking that these values above are very broad ranges. How can you really know how many reads you need? The short answer is you don’t. You can use other publications as a guide and you can talk to other genomics people, but it is often an approximation. This section is here to give you a guide on how you can make a pretty good approximation of what you will need!
The next thing the sequencing facility will ask you if you are doing short-read sequencing (Illumina) is: do you want single-end or paired-end reads? This refers to whether you want a single read (read 1), sequenced from only one end of the library molecule (fragment), or if you want two reads per library molecule (read 1 and read 2, antisense and sense strands). Paired-end costs more, but gives you more resolution.
There are pros and cons to choosing either chemistry:
Table 2: Single-end vs paired-end chemistry (short-read Illumina sequencing)
| Chemistry | Pros | Cons |
|---|---|---|
| Single-end (SE) | Lower cost, fewer reads required (may be able to do more samples); sufficient for basic gene-level DGE | Limited splice/isoform resolution; less confident mapping when mapping to a genome |
| Paired-end (PE) | Better alignment; improved splice junction and isoform detection; more robust for complex transcriptomes or de novo transcriptome assembly | Higher cost; ~2× sequencing required (two reads per fragment) |
Lastly, if you are doing short-read sequencing (i.e., Illumina), the sequencing facility will ask you what read length you want. You can typically chose from between 50bp-300bp (platform-dependent). The choice between a lower or higher read length will be a balance of cost (higher read length = higher cost) and the level of information you need (complex, novel or de novo transcriptomes require higher read lengths; more straight forward analyses with well-annotated genomes can utilise lower read lengths). In contrast, for long-read sequencing platforms (ONT and PacBio), you do not specify a fixed read length. Instead, reads are generated as single, continuous sequences, and their length is determined by the size of the input molecules, the library preparation method, and the sequencing chemistry.
Choosing a read length is a trade-off between cost, and the amount of information you will recover:
Table 3: Read length (short-read Illumina sequencing)
| Read length | Pros | Cons |
|---|---|---|
| 50 bp | Lowest cost; highest sample multiplexing; sufficient for basic gene-level DGE in well-annotated genomes | Poor isoform and splice junction resolution; higher multi-mapping |
| 100 bp | Good balance of cost and information; reliable splice junction detection; widely used for standard RNA-seq | Slightly lower throughput than 50 bp; may miss very complex isoforms |
| 150 bp | Improved isoform resolution; better mapping across repetitive regions; useful for novel transcript discovery | Higher cost; fewer reads per run |
| 300 bp | Maximum per-read information; helpful for de novo transcriptome assembly | Rarely necessary for Illumina RNA-seq; expensive; reduced throughput |
Congratulations! You are now ready to sequence your RNAseq samples.
Now let’s look at DNA sequencing in more detail.
DNA sequencing may refer to whole genome, whole exome, or targeted sequencing approaches.
more explanation by someone who knows DNAseq better!
For Genome sequencing you will need:
| Genome size | Example species | Approx genome size | Approx reads per sample |
|---|---|---|---|
| Tiny | Virus | <0.1 Mb | 0.1–0.5 million |
| Small | Bacteria | 5 Mb | 5–10 million |
| Medium | Yeast | 12 Mb | 10–20 million |
| Large | Fruit fly (D. melanogaster) | 175 Mb | 50–100 million |
| Very Large | Human | 3 Gb | 600–1,200 million |
| Huge | Wheat | 16 Gb | 6–12 billion |
Where Mb = megabase pairs (i.e., 1 Mb = 1,000,000 bp)
Coverage. talking about average coverage - really repetitive regions can have very low covergae, other more easily resolved sections will have higher coverage.
heterozygosity homozygosity.
haplotypes etc.
Stick mostly to decision points - not a huge lecture/tutorial on genome/ DNA biology. Assume learners haev a genetics background - they just have not yet translated that knowledge into a practical application of NGS.
Note 1: Batch variation. Are you doing all your samples in one batch, or will you have multiple batches? Technical variation can occur, so you may want to wait and do all samples at once on one larger flow cell, or make sure you randomise samples across sequencing batches.
Note 2: DNAseq vs RNAseq. You may have noticed we call it ‘RNAseq’, even though we convert the RNA into DNA before sequencing! By convention, this is still called RNAseq, to differentiate it from true DNAseq. ONT can directly sequence the RNA without converting it to DNA first; we call this native RNA sequencing.
NZ sequencing facilities and services
Current as of: January 2026
not planning to go into detail at all on non-NGS services around NZ. Sticking to high throughput genomics.
There are several dedicated sequencing facilities which offer NGS services around New Zealand that you can use. Most accept samples from any researcher, but there may be a higher cost for non-staff or students.
Otago Genomics Facility (OGF)
Location: The University of Otago, Biochemistry Building, 710 Cumberland st.
Performs: Moderate to large scale RNAseq, DNAseq, amplicon sequencing, other sequencing (Illumina library prep and sequencing provided as a service; ONT platform requires the users to undertake the library and sequencing themselves with supported training and trouble-shooting advice).
Website: Otago Genomics Facility
Platforms available:
- Illumina NextSeq 2000
- Illumina MiSeq
- ONT P2 Solo
- ONT MinION
- NanoString nCounter Analysis System (non-NGS)
Massey Genome Service (MGS)
Location: Massey University, ADDRESS
Performs: Small scale RNA or DNAseq, single gene (Sanger) sequencing.
Website: Massey Genome Service
Platforms available:
- 2 x Illumina MiSeq
- Applied Biosystems 3500xl capillary instrumentation (non-NGS).
Notes:
- Offers the Illumina TruSeq library prep method
Auckland Genomics
Location: ADDRESS
Performs: FILL
Website: Auckland Genomics
Platforms available:
- Illumina FILL
- ONT FILL
Notes:
- Offers the Illumina Stranded mRNA library prep method
Other services and platforms around New Zealand:
GenomNZ at AgResearch, Invermay are a commercial animal DNA genotyping laboratory, primarily for sheep, cattle, deer, goat and aquaculture sequencing and use an Illumina NovaSeq.
Lincoln have a MGI DNBSEQ-G400 genome sequencer, which is compatible with Illumina libraries. Unclear how or if researchers can use this
Custom science (a supplier, does not do the sequencing for you) have negotiated the installation of a new PacBio Revio in Auckland
Bragato Research Institute are a grape and wine research institute in Blenheim, and have an ONT PromethION Sequencer which is available as a service to other research agencies and customers.
Grafton Clinical Genomics perform Illumina and Ion Torrent next-generation sequencing, to support research, clinical and translational groups at the University of Auckland and Auckland DHB as part of the Academic Health Alliance relationship.
Working with NZ sequencing facilities
stuff on submission process, show example form and brief explanation of how to fill out? I might have covered enough of this anywya by explaing libraries and reads etc.
how do you get data back from them ? how do they transfer it?
Do all the seq facilities in NZ give you data back demultiplexed by default?
Do anyone use basespace to share reads with people?
International sequencing services
International sequencing services - touch on this, why you may chose over NZ (or not chose!) then point towards the next section on ethical data management.
Shipping and storage logistics:
- RNA stability, and the need to preserve RNA (by freezing or other) for overseas sequencing?
- silicon tubes that will keep the RNA good and reduce sequencing costs